
ę╗Īó╗∙▒Šą┼Žó
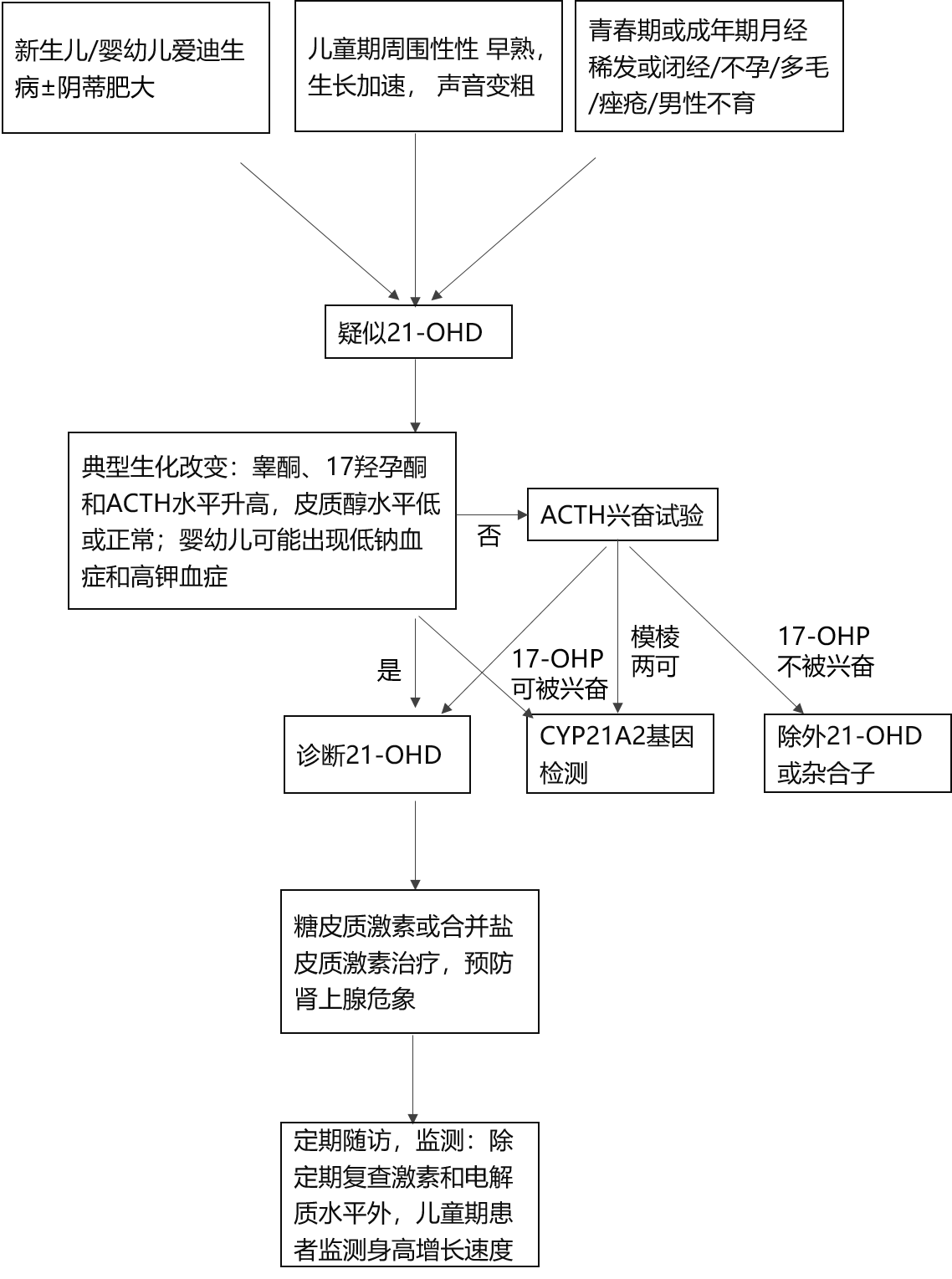
Ė┼╩÷Ż║21-┴u╗»├Ė╚▒Ę”░YŻ©21-hydroxylase deficiencyŻ¼21-OHDŻ®╩ŪŽ╚╠ņąį─I╔ŽŽ┘į÷╔·░YŻ©congenital adrenal hyperplasiaŻ¼CAHŻ®ųąūŅ│ŻęŖĄ─ŅÉą═Ż¼╩Ūė╔ė┌ŠÄ┤a21-┴u╗»├ĖĄ─CYP21A2╗∙ę“╚▒Ž▌ī¦ų┬─I╔ŽŽ┘Ųż┘|ŅÉ╣╠┤╝╝ż╦ž║Ž│╔šŽĄKĄ─ę╗ĘNŽ╚╠ņąį╝▓▓ĪŻ¼│╩│Ż╚Š╔½¾wļ[ąį▀zé„ĪŻĮøĄõą═╗╝š▀┐╔░l╔·─I╔ŽŽ┘╬ŻŽ¾Ż¼ī¦ų┬╔·├³╬ŻļUŻ╗Ė▀ą█╝ż╦žč¬░Y╩╣┼«ąį─ąąį╗»Ż¼ī¦ų┬╣Ū²g╝ė╦┘▀Mš╣Īó░½╔Ē▓─ęį╝░ŪÓ┤║░lė²«É│ŻŻ¼▓óė░Ēæ╔·ė²─▄┴”ĪŻ
▓Īę“Ż║21-OHDė╔╬╗ė┌╚Š╔½¾w6p21.3ģ^ė“ā╚Ą─CYP21A2╗∙ę“═╗ūāę²ŲĪŻŲõŠÄ┤aĄ─Ą░░ū×ķ21┴u╗»├ĖŻ©P450c21Ż®ĪŻįō├Ė┤▀╗»17┴uįą═¬Ż©17-OHPŻ®▐D╗»×ķ11-├ōč§Ųż┘|┤╝Ż¼═¼Ģr┤▀╗»įą═¬▐D╗»×ķ11-├ōč§Ųż┘|═¬Ż¼Č■š▀Ęųäe×ķŲż┘|┤╝║═╚®╣╠═¬Ą─Ū░¾wĪŻ21┴u╗»├Ė╗ŅąįĮĄĄ═ų┬Ųż┘|┤╝║═╚®╣╠═¬║Ž│╔╩▄ōpĪŻŲż┘|┤╝╦«ŲĮ║Ž│╔£p╔┘Ż¼═©▀^žōĘ┤ü╩╣┤╣¾wACTHĘų├┌į÷╝ėŻ¼┤╠╝ż─I╔ŽŽ┘Ųż┘|╝Ü░¹į÷╔·Ż╗Č°╚®╣╠═¬Ęų├┌▓╗ūŃ╝ż╗Ņ╔Žė╬─I╦ž║═č¬╣▄ŠoÅł╦žó“Ą─Ęų├┌Ż¼═¼Ģrė╔ė┌ųąķg«a╬’Ą─ČčĘeŻ¼×ķąį╝ż╦žŻ©į┌─I╔ŽŽ┘Ųż┘|ų„ę¬×ķą█╝ż╦žŻ®║Ž│╔╠ß╣®┴╦«É│Żį÷ČÓĄ─Ąū╬’Ż¼«a╔·┴╦┼į┬Ę┤·ųx┐║▀MĄ─╠žš„ąį║¾╣¹Ī¬Ī¬Ė▀ą█╝ż╦žč¬░YĪŻą█╝ż╦ž╔²Ė▀’@ų°│╠Č╚ę└┤╬×ķą█Ž®Č■═¬Īó▓G═¬║═├ōÜõ▒Ēą█═¬Ż©DHEAŻ®ĪŻ
CYP21A2╗∙ę“Ą─═╗ūāŅÉą═ėą░┘ėÓĘNŻ¼80Żź┤µį┌╗∙ę“ą═║═▒Ēą═Ą─ŽÓĻPąįĪŻ«ö═╗ūāī¦ų┬21┴u╗»├Ė╗ŅąįĄ═ė┌1ŻźĢrŻ¼▒Ē¼F×ķć└ųž╩¦¹}Ż¼│╩¼FĄ═Ōcč¬░Y║═Ė▀Ōøč¬░YŻ¼ą┬╔·ā║─I╔ŽŽ┘╬ŻŽ¾ĪŻ«ö├Ė╗ŅąįÜł┴¶×ķ1ŻźĪ½2ŻźĢrŻ¼╚®╣╠═¬▀Ć┐╔ŠS│ųį┌š²│ŻĘČć·Ż¼╩¦¹}āAŽ“Ą═Ż©Ą½æ¬╝żĢr╚į┐╔░l╔·Ż®ĪŻ├Ė╗Ņąį▒Ż┴¶ėą20ŻźĪ½50ŻźĢrŻ¼Ųż┘|┤╝║Ž│╔Äū║§▓╗╩▄ōpĪŻ░┤╗∙ę“ą═-┼R┤▓▒Ēą═Ą─ĻPŽĄĪó╚®╣╠═¬║═Ųż┘|┤╝╚▒Ę”Ą─│╠Č╚ĪóĖ▀ą█╝ż╦žĄ─ć└ųž│╠Č╚Ż¼21-OHDĘų×ķā╔┤¾ŅÉą═Ż║
1.ĮøĄõą═21-OHDŻ¼░┤╚®╣╠═¬╚▒Ę”│╠Č╚ėųĘų×ķ╩¦¹}ą═Ż©salt wastingŻ¼SWŻ¼╝sš╝75ŻźŻ®║═å╬╝ā─ąąį╗»ą═Ż©simple virilizingŻ¼ SVŻ¼╝sš╝25ŻźŻ®Ż╗
2.ĘŪĮøĄõą═21-OHDŻ©non classical CAHŻ¼NCCAHŻ®ĪŻ
┴„ąą▓ĪīWŻ║CAHĖ∙ō■╚▒Ž▌├ĖĄ─ĘNŅÉ┐╔Ęų×ķČÓĘNŅÉą═Ż¼21-OHD╩ŪūŅ│ŻęŖĄ─ŅÉą═Ż¼š╝90ŻźĪ½95ŻźĪŻć°ā╚═ŌĄ─ČÓöĄčąŠ┐╠ß╩Šą┬╔·ā║║Y▓ķ░l▓Ī┬╩×ķ1/20000Ī½1/10000ĪŻ
Č■Īó╝▓▓Īį\öÓ
┼R┤▓▒Ē¼FŻ║21-OHDĄ─┼R┤▓▒Ē¼F░³└©▓╗═¼│╠Č╚Ą─╩¦¹}║═Ė▀ą█╝ż╦žč¬░Yā╔┤¾ŅÉĪŻą┬╔·ā║Ų▓ĪĄ─╗╝ā║▒Ē¼F×ķ▓╗═¼│╠Č╚─I╔ŽŽ┘Ųż┘|╣”─▄▓╗ūŃĄ─▒Ē¼FŻ¼╚ń▄ø╚§¤o┴”ĪóÉ║ą─ĪóćI═┬Īó╬╣B└¦ļyĪóĖ╣×aĪó┬²ąį├ō╦«ĪóŲż─w╔½╦ž│┴ų°║═╔·ķL▀tŠÅĄ╚ĪŻ─I╔ŽŽ┘╬ŻŽ¾│Ż╩Ū21-OHD SWą═į┌ą┬╔·ā║Ų┌Ą─╩ū░l▒Ē¼FŻ¼▒Ē¼F×ķć└ųžĄ═č¬ŌcĪóĖ▀č¬ŌøĪóĄ═č¬╚▌┴┐ąįą▌┐╦Ż¼┐╔░ķėąĄ═č¬╠ŪŻ¼ė╔æ¬╝żšT░lĪŻć└ųžĄ─Ą═Ōcč¬░Y┐╔ī¦ų┬│ķ┤żĄ╚ųąśą╔±ĮøŽĄĮy▒Ē¼FŻ¼ć└ųžĄ─Ė▀Ōøč¬░Yät┐╔ę²Ųų┬├³Ą─ą─┬╔╩¦│ŻĪŻ
Ė▀ą█╝ż╦žč¬░Yį┌▓╗═¼─Ļ²g▒Ē¼F▓╗ę╗ĪŻĮøĄõą═21-OHDĄ─┼«ąį╗╝ā║│÷╔·Ģr═Ō╔·ų│Ų„ėą▓╗═¼│╠Č╚─ąąį╗»ĪŻ▌pš▀▒Ē¼F×ķ╣┬┴óąįĻÄĄ┘Ę╩┤¾Ż¼ųžš▀═Ō╔·ų│Ų„┐╔ĮėĮ³─ąąįŻ¼┤¾ĻÄ┤Į│╩ĻÄ─ęśėŻ¼ĻÄĄ┘│╩─“Ą└Ž┬┴čą═ĻÄŪośėŻ¼▓óŠ▀╣▓═©Ą──“Ą└ĻÄĄ└┐┌ĪŻĄ½┤¾ĻÄ┤Įā╚▓╗─▄ė|╝░ąįŽ┘Ż¼ėą═Ļ╚½š²│ŻĄ─┼«ąįā╚╔·ų│Ų„ĮYśŗŻ©┬č│▓║═ūėīmŻ®ĪŻ─ąąįą┬╔·ā║Ų┌║═ŗļā║Ų┌Ģr┐╔─▄¤oĻÄŪoį÷┤¾Ą╚═Ō╔·ų│Ų„«É│ŻŻ¼╩Ūčėš`į\öÓĄ─│ŻęŖįŁę“ĪŻų┴ėūā║Ų┌Ż¼ā╔ąįŠ∙Ģ■│╩¼F═Ōų▄ąįąįįń╩ņĪŻ─ą║ó╗╝ā║│╩¼FĻÄŪoį÷┤¾Ż¼░ķ╗“▓╗░ķĻÄ├½įń¼FŻ╗┼«ąį╗╝ā║│╩¼F«Éąįąįįń╩ņĪŻķLŲ┌Ė▀╦«ŲĮąį╝ż╦ž┤╠╝żŽ┬Ū─X┤┘ąįŽ┘╝ż╦žßīĘ┼╝ż╦žŻ©GnRHŻ®╔±Įøį¬Ż¼░lš╣×ķųąśąąįąįįń╩ņĪŻ┼«ąį▀Ć┐╔ėąĄ┌Č■ąįš„░lė²▓╗┴╝║═įŁ░ląįķ]Įø╗“į┬ĮøŽĪ░lĪŻ21-OHDĘŪĮøĄõą═į┌ŪÓ┤║Ų┌╗“│╔─Ļ║¾▒╗öMį\×ķČÓ─ę┬č│▓ŠC║Žš„Ģr▓┼▒╗┤_į\×ķ21-OHDĪŻā╔ąįŠ∙į┌ėū─ĻŲ┌ķ_╩╝░l╔·ŠĆąį╔·ķL░ķ╣Ū²gį÷ķL╝ė╦┘Ż¼╩╣ĮK╔ĒĖ▀╩▄ōpĪŻ
Ųõ╦¹▒Ē¼F░³└©Ųż─w║═ż─ż╔½╦žį÷╔ŅŻ¼ęį╚ķĢ×║══ŌĻÄ×ķ’@Ż¼▓┐Ęų╗╝ā║┐╔¤o┤╦Ė─ūāĪŻ
NCCAHā║═»Ų┌║═ŪÓ┤║Ų┌╔§ų┴│╔─ĻŲ┌┼R┤▓│╩▓╗═¼│╠Č╚Ą─Ė▀ą█╝ż╦žč¬░Y▒Ē¼FŻ¼ę▓ėąāH▒Ē¼F×ķ╔·ķL╝ė╦┘║═╣Ū²g┐ņ╦┘▀Mš╣ĪŻ
▌oų·Öz▓ķŻ║
1.č¬ŪÕ17-OHP
17-OHP╔²Ė▀╩Ū21-OHDĄ─╠ž«Éąįį\öÓųĖś╦║═ų„ę¬ų╬»¤▒O£yųĖś╦ĪŻę╗░ŃČ°čįŻ¼17-OHP╔²Ė▀Ę∙Č╚įĮĖ▀Ż¼├Ė╚▒Ž▌│╠Č╚įĮųžĪŻĄ½17-OHP┼cACTHę╗śėČ╝╩Ūæ¬╝ż╝ż╦žŻ¼ę“┤╦į┌ėąæ¬╝żĄ─ŪķørŽ┬Ż¼╗“╗╝ā║│ķč¬┐▐¶[ć└ųžĢrČ╝┐╔▌^īŹļH╦«ŲĮėą├„’@Ą─╔²Ė▀Ż¼ĮŌūxĮY╣¹ĢrąĶ┐╝æ]ĄĮ┤╦ĘNę“╦žĪŻ
2.╗∙ĄAč¬ŪÕŲż┘|┤╝║═ACTH
ĮøĄõą═╗╝š▀č¬ŪÕŲż┘|┤╝ĮĄĄ═░ķACTH╔²Ė▀ĪŻę▓ėą21-OHD╗╝š▀Ųż┘|┤╝į┌š²│ŻĘČć·Ż¼Č°ACTH╔²Ė▀Ż¼ąĶĮY║ŽŲõ╦¹ųĖś╦ŠC║Ž┼ąöÓĪŻ21-OHDĘŪĮøĄõą═╗╝š▀ā╔ĘN╝ż╦ž╗∙▒Šį┌š²│ŻĘČć·ĪŻ
3.ą█╝ż╦ž
Ė„ą█╝ż╦ž£yČ©ųĄąĶ░┤ššąįäeĪó─Ļ²g║═ŪÓ┤║░lė²Ų┌Į©┴óĄ─š²│ŻģóššųĄ┼ąöÓĪŻą█Ž®Č■═¬┼c17-OHPėą▌^║├Ą─ŽÓĻPąįŻ¼į\öÓ║═▒O£yęŌ┴xūŅ╝čĪŻDHEASį┌ČÓ─ę┬č│▓ŠC║Žš„ųąęÓ┐╔ėą╔²Ė▀ĪŻ
4.č¬Ø{─I╦žØŌČ╚╗“─I╦ž╗ŅąįĪóč¬╣▄ŠoÅł╦žó“║═╚®╣╠═¬
─I╦žį┌21-OHD SWą═╔²Ė▀Ż¼Ą½į\öÓ╠ž«Éąį▓╗Ė▀ĪŻ▓┐ĘųĘŪ╩¦¹}ą═╗╝š▀─I╦žę▓┐╔╔²Ė▀Ż¼─I╦ž╩Ū¹}Ųż┘|╝ż╦ž╠µ┤·ų╬»¤ųąĄ─ųžę¬▒O£yųĖś╦ĪŻ╚®╣╠═¬Ą═Ž┬ų¦│ų╩¦¹}ą═į\öÓŻ¼Ą½ėą1/4╗╝ā║č¬ŪÕ╚®╣╠═¬┐╔š²│ŻĪŻ
5.╚Š╔½¾w
╚Š╔½¾wų„ę¬ė├ė┌│²═Ō46Ż¼XYąį░lė²«É│Ż╝▓▓ĪĪŻ
6.ė░Ž±īW
─I╔ŽŽ┘Ą─B│¼║═CTĄ╚ė░Ž±īWÖz▓ķėąų·ė┌─I╔ŽŽ┘─[┴÷╗“Ųõ╦¹─I╔ŽŽ┘Ż©░lė²▓╗┴╝Ż®▓ĪūāĶbäeĪŻ┼«ąįæ¬═Ļ╔ŲūėīmĪóļpĖĮ╝■B│¼Ż¼2Üqķ_╩╝ąĶÖz▓ķ╣Ū²gĪŻ
7.╗∙ę“į\öÓ
21-OHDĄ─╗∙ę“į\öÓ¤ošōī”╔·╗»į\öÓ├„┤_╗“š▀▓╗├„┤_Ą─Ķbäeį\öÓČ╝╩«Ęųųžę¬Ż¼▓óŪę─▄į\öÓļs║ŽūėöyĦš▀Ż¼ī”▀zé„ū╔įāę▓ĘŪ│Żųžę¬ĪŻ
į\öÓŻ║21-OHDį\öÓąĶŠC║Ž┼R┤▓▒Ē¼FĪó░³└©17-OHPį┌ā╚Ą─Ė„ŽÓĻP╝ż╦žØŌČ╚üĒ╝ėęį┼ąöÓŻ¼╗∙ę“Öz£y┐╔▀Mę╗▓Į├„┤_į\öÓĪŻ─┐Ū░ć°ā╚▌^ČÓĄžģ^ęčķ_š╣21-OHDĄ─ą┬╔·ā║║Y▓ķŻ¼ī”ė┌ūŃĖ·č¬║Y▓ķ17-OHPĻ¢ąįš▀Ż¼ąĶ░┤šš║Y▓ķ╣▓ūR▓┘ū„ĪŻ
NCCAH╗╝š▀č¬ŪÕŲż┘|┤╝š²│Ż╗“į┌š²│ŻŽ┬Ž▐Ż¼ACTHš²│Ż╗“┼RĮńĖ▀ųĄĪŻė├17-OHP╗∙ĄAųĄį\öÓŠ▀▓╗┤_Č©ąįŻ¼╗∙ę“Öz£yśOŲõųžę¬ĪŻ
Ķbäeį\öÓŻ║│ŻęŖĄ─ąĶę¬┼c21-OHDĶbäeį\öÓĄ─╝▓▓Ī░³└©Ż║
1.11”┬-┴u╗»├Ė╚▒Ž▌Ż©CYP11B1╗∙ę“═╗ūāŻ®
ę▓ėąĖ▀ą█╝ż╦žč¬░YŻ¼śO╔┘ėą│÷╔·Ģr╩¦¹}▒Ē¼FŻ¼│ŻęŖ¹}Ųż┘|╝ż╦ž▀^ČÓ╚ń╦«Ōcõ¾┴¶ĪóĄ═č¬Ōø║═Ė▀č¬ē║Ą╚Ż¼─I╦ž-č¬╣▄ŠoÅł╦ž╦«ŲĮĄ═Ż¼įą═¬┼c17-OHP╔²Ė▀ĪŻĄ½▓┐Ęų╗╝š▀č¬ē║┐╔š²│ŻŻ¼▒žę¬ĢrąĶąą╗∙ę“Öz£y┼c21-OHDĶbäeĪŻ
2.17”┴-┴u╗»├Ė╚▒Ž▌░YŻ©CYP17A1╗∙ę“═╗ūāŻ®
┤╦├Ė═¼Ģr▀ĆŠ▀ėą17Ż¼20-┴čµ£├ĖĄ─╗ŅąįŻ¼┼R┤▓▒Ē¼F×ķ¹}Ųż┘|╝ż╦žį÷ČÓĄ─░YĀŅŻ¼╚ńĄ═č¬ŌøĪóĖ▀č¬ē║ęį╝░ąį╝ż╦ž▓╗ūŃĄ─▒Ē¼FŻ¼╚ń┼«ąįŪÓ┤║░lė²╚▒╩¦Ż¼─ąąį┼«ąį╗»ĪŻįą═¬╔²Ė▀Ż¼17-OHPĮĄĄ═╗“š²│ŻĪŻ
3.Ž╚╠ņąį▀zé„ąį─I╔ŽŽ┘░lė²▓╗┴╝
╩Ūė╔ė┌NR0B1╗∙ę“╗“SF1╗∙ę“═╗ūāī¦ų┬Ą─Ž╚╠ņąį─I╔ŽŽ┘Ųż┘|╣”─▄£p═╦Ż¼┐╔║Ž▓óąįŽ┘╣”─▄Ą═Ž┬Ż¼Ųõė░Ž±īWČÓ▒Ē¼F×ķ─I╔ŽŽ┘░lė²▓╗┴╝ĪŻ
4.─I╔ŽŽ┘Ųż┘|─[┴÷
─I╔ŽŽ┘Ųż┘|─[┴÷Ż©ė╚Ųõ╩Ūā║═»Ż®│ŻęįĖ▀ą█╝ż╦žč¬░YĄ─┼R┤▓▒Ē¼FŲ▓ĪŻ¼░ķ╗“▓╗░ķŲż┘|┤╝į÷ČÓ░YŻ¼╔§ų┴ėą17-OHP’@ų°╔²Ė▀Ż¼Ą½ACTH├„’@Ą═Ž┬╩ŪĶbäeę¬³cĪŻė░Ž±īWūCīŹš╝╬╗▓ĪūāĪŻ
5.ČÓ─ę┬č│▓ŠC║Žš„
ī”ė┌ŪÓ┤║Ų┌╗“│╔─ĻŲ┌ę“į┬Įø╩¦š{╗“Ė▀ą█╝ż╦žč¬░YŠ═į\Ą─┼«ąį╗╝š▀Ż¼NCCAHĄ─▒Ē¼F┐╔┼cČÓ─ę┬č│▓ŠC║Žš„ėąę╗Č©ųž»BŻ¼ŪęČÓ─ę┬č│▓ŠC║Žš„ęÓ┐╔│÷¼FDHEASĄ─╔²Ė▀Ż¼┐╔═©▀^ųąä®┴┐Ąž╚¹├ū╦╔ęųųŲįć“×ĶbäeŻ¼▒žę¬ĢrąąCYP21A2╗∙ę“Öz£yęį├„┤_į\öÓĪŻ
╚²Īóų╬»¤ĘĮ╩Į
ų╬»¤Ż║
1.ų╬»¤─┐ś╦
░┤šš21-OHD▓╗═¼ŅÉą═ųŲČ©ų╬»¤─┐ś╦ĪŻų╬»¤─┐ś╦░³└©╠µ┤·╔·└ĒąĶę¬┴┐Ą─╠ŪŲż┘|╝ż╦žŻ¼═¼Ģr║Ž└ĒęųųŲĖ▀ą█╝ż╦žč¬░YŻ¼▒M┐╔─▄╗ųÅ═š²│Ż╔·ķL░lė²Ą─▄ē█EŻ¼▀_ĄĮ└ĒŽļĄ─ĮK╔ĒĖ▀Ż¼Ė─╔Ų▀hŲ┌╔·ų│ĮĪ┐ĄĪŻ
2.╠ŪŲż┘|╝ż╦žų╬»¤
Üõ╗»┐╔Ą─╦╔╩Ū╗∙ĄAė├╦ÄŻ¼ąĶę¬ĮK╔·Ą─╠µ┤·ų╬»¤ĪŻĮ©ūhĘųäe░┤šš╗╝š▀╔ąį┌╔·ķLųą║═ęč▀_ĄĮ│╔─Ļ╔ĒĖ▀ŪķørųŲČ©ĘĮ░ĖĪŻ╬┤═Żų╣╔·ķLš▀Ż¼Į©ūhė├Üõ╗»┐╔Ą─╦╔╠µ┤·ĪŻ▀_ĄĮ│╔─Ļ╔ĒĖ▀║¾Ż¼┐╔ęįĮo░ļ╦źŲ┌ŽÓī”ķLĄ─ųŲ䮯¼╚ńØŖ─ß╦╔╗“Ąž╚¹├ū╦╔ĪŻÜõ╗»┐╔Ą─╦╔╠µ┤·ų╬»¤ĘĮ░ĖąĶį┌ģóšš╦Ä╬’┤·ųxäė┴”īWĄ─įŁät╔ŽĮ©┴óéĆ¾w╗»ĘĮ░ĖĪŻĖ∙ō■─Ļ²gįOČ©ä®┴┐Ż¼Ęų┤╬Įo╦ÄŻ¼Ė∙ō■▒O£y▀Mąąä®┴┐š{╣ØĪŻ╠µ┤·ų╬»¤ä®┴┐║═ĘĮ░Ėæ¬ĮY║Ž─Ļ²g║═░lė²Ų┌éĆ¾w╗»įOČ©Ż¼▓ó▒M┐╔─▄┐žųŲį┌ūŅĄ═ėąą¦ä®┴┐Ż¼▒▄├Ō▀^┴┐ī”╔·ķLĄ─ęųųŲ║═░l╔·ßtį┤ąįÄņą└ŠC║Žš„ĪŻį┌æ¬╝żĀŅæB║═╝▓▓ĪĢrąĶī”╠ŪŲż┘|╝ż╦žĄ─ä®┴┐▀Mąąš{š¹ĪŻę╗░Ńā║═»ė├┴┐Üõ╗»┐╔Ą─╦╔10Ī½15mg/m2Ż¼├┐╚šĘų3┤╬┐┌Ę■Ż╗│╔╚╦ė├┴┐15Ī½25mg/dŻ¼├┐╚šĘų2Ī½3┤╬┐┌Ę■ĪŻ
3.¹}Ųż┘|╝ż╦žų╬»¤
ąĶę¬į┌Ę└ų╣╩¦¹}╬ŻŽ¾═¼ĢrŻ¼ĻPūó¹}╝ż╦ž├¶ĖąąįĄ──Ļ²gūā╗»ęÄ┬╔╝░Ģrš{š¹ä®┴┐Ż¼▒▄├Ō░l╔·ßtį┤ąįĖ▀č¬ē║ĪŻ21-OHD╩¦¹}ą═į┌╠ŪŲż┘|╝ż╦ž╗∙ĄA╔Ž┬ōė├¹}Ųż┘|╝ż╦ž┐╔ęį£p╔┘╠ŪŲż┘|╝ż╦žĄ─┐é┴┐╝░ķLŲ┌▓╗┴╝Ę┤æ¬ĪŻĘ·Üõ┐╔Ą─╦╔╩Ū─┐Ū░╬©ę╗Ą─¹}Ųż┘|╝ż╦žųŲ䮯¼┐╔ęį├┐╚šĘų1Ī½2┤╬Ę■ė├Ż¼ä®┴┐░┤ī”¹}Ųż┘|╝ż╦ž├¶ĖąąįĄ──Ļ²gĖ─ūāęÄ┬╔įOų├Ż¼Ė∙ō■▒O£y▀Mąąä®┴┐š{╣ØĪŻė╔ė┌ų┴Į±╔ą╬┤ėą┼ąöÓ»¤ą¦Ą─å╬ę╗╝ż╦žęį╝░╣╠Č©Ą─Ūą³cĄ─Ī░Įś╦£╩Ī▒Ż¼Į©ūhąĶĮY║Ž╝ż╦ž║═┼R┤▓ųĖś╦▒O£yŠC║Ž┼ąöÓŻ¼īŹ¼F▀_ĄĮéĆ¾w╗»ų╬»¤Ą─ūŅ└ĒŽļ─┐ś╦ĪŻę╗░Ńā║═»ė├┴┐Ę·Üõ┐╔Ą─╦╔0.05Ī½0.2mg/dŻ¼├┐╚šĘų1Ī½2┤╬Ę■ė├Ż╗│╔╚╦ä®┴┐0.05Ī½0.2mg/dŻ¼├┐╚š1┤╬┐┌Ę■ĪŻ
4.╔·ķL╝ż╦ž║═┤┘ąįŽ┘╝ż╦žßīĘ┼╝ż╦žŅÉ╦Ų╬’
ī”ė┌ąįįń╩ņć└ųžŻ¼╣Ū²g│¼Ū░├„’@Ż¼ŅA£y│╔─Ļ╔ĒĖ▀ōp╩¦▌^ČÓš▀Ż¼┐╔┐╝æ]╔·ķL╝ż╦žų╬»¤ĪŻī”ė┌ęčĮø░l╔·ųąśąąįąįįń╩ņĄ─╗╝š▀Ż¼┐╔┬ō║Ž┤┘ąįŽ┘╝ż╦žßīĘ┼╝ż╦žŅÉ╦Ų╬’ĪŻĄ½┤╦ŅÉ╦Ä╬’ī”ė┌ĮK╔ĒĖ▀Ą─Ė─╔Ų┼c╗╝š▀įŁ░l▓ĪĄ─┐žųŲĪóī”╦Ä╬’Ą─ų╬»¤Ę┤æ¬Īóī”╣Ū²g│¼Ū░Ą─│╠Č╚ĪóĖĖ─ĖĄ─▀zé„╔ĒĖ▀Ą╚ČÓĘĮ├µę“╦žėąĻPŻ¼ę“┤╦ų╬»¤ą¦╣¹éĆ¾w▓Ņ«É▌^┤¾ĪŻ
į\»¤┴„│╠łDŻ║
ģó┐╝╬─½IŻ║
[1] Speiser PW,Azziz R,Baskin LS,et al.Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab,2010,95(9):4133-4160.
[2] Fleming L,Van Riper M,Knafl K. Management of childhood congenital adrenal hyperplasia-an integrative review of the literature. J Pediatr Health Care,2017,31(5):560-577.
[3] Anzo M,Adachi M,Onigata K,et al. Guidelines for diagnosis and treatment of 21-hydroxylase deficiency (2014 revision).Clin Pediatr Endocrinol,2015,24(3):77-105.
[4] ┤„║├, ▒R┴š, ąŽąĪŲĮ, Ą╚. ųąä®┴┐Ąž╚¹├ū╦╔ą█╝ż╦žęųųŲįć“×į┌┼«ąįĖ▀ą█╝ż╦žč¬░YųąĄ─į\öÓārųĄ. ųą╚AßtīWļsųŠ,2018,98(26):2073-2077.
[5] ųą╚AßtīWĢ■ā║┐ŲīWĘųĢ■ā╚Ęų├┌▀zé„┤·ųx▓ĪīWĮM. Ž╚╠ņąį─I╔ŽŽ┘Ųż┘|į÷╔·░Y21-┴u╗»├Ė╚▒Ž▌į\ų╬╣▓ūR. ųą╚Aā║┐ŲļsųŠ, 2016,54(8):569-576.
░µÖÓ┬Ģ├„
ęį╔Žā╚╚▌üĒūį┴╝ßtģR-║▒ęŖ▓Īą┬▀Mš╣Ż¼╚ńėąĮ©ūh╗“ę╔å¢Ż¼ÜgėŁų┬ļŖ18017449015ĪŻ
 ķL░┤Č■ŠS┤a
ķL░┤Č■ŠS┤aĻPūóŠ½▓╩ā╚╚▌





