
ę╗Īó╗∙▒Šą┼Žó
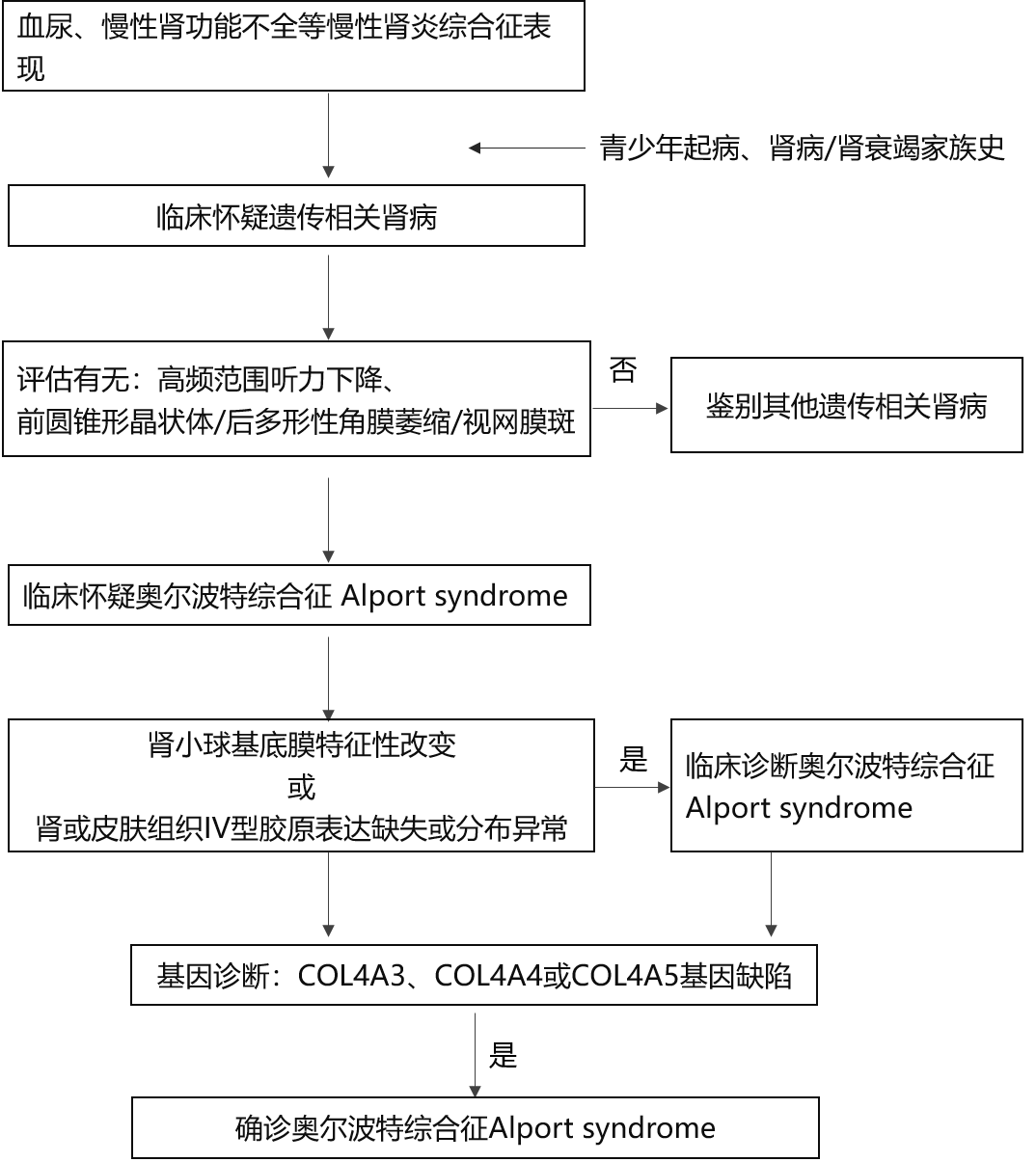
Ė┼╩÷Ż║ŖWĀ¢▓©╠žŠC║Žš„Alport syndromeŻ©▒Š╬─║¾ĘQAlportŠC║Žš„Ż®╩Ūę╗ĘN▀zé„ąį─zįŁ▓ĪŻ¼×ķŠÄ┤aó¶ą═─zįŁĄ░░ū”┴-3µ£Īó”┴-4µ£║═”┴-5µ£Ą─╗∙ę“COL4AnŻ©n=3Īó4Īó5Ż®═╗ūāī¦ų┬Ą─╗∙Ąū─ż▓ĪūāĪŻūŅ│Ż└█╝░─I┼KŻ¼Ųõ┤╬└█╝░č█║═Č·ĪŻė╔ė┌╔Ž╩÷═╗ūāī¦ų┬Ų„╣┘─zįŁĮY(ji©”)śŗ(g©░u)«É│ŻŻ¼▀MČ°▒Ē¼F(xi©żn)×ķ▀zé„ąį─IąĪŪ“╝▓▓ĪŻ¼░³└©č¬─“ĪóĄ░░ū─“╝░─I╣”─▄▀MąąąįŽ┬ĮĄŻ¼│Ż░ķėąĖąę¶╔±Įø(j©®ng)ąį┬Ā┴”ōp╩¦║═č█▓┐«É│ŻĪŻ
▓Īę“Ż║AlportŠC║Žš„▓Ī└ĒÖCųŲĄ─Ęųūė╗∙ĄA(ch©│)╩ŪŠÄ┤aó¶ą═─zįŁĄ░░ūĄ─╗∙ę“░l(f©Ī)╔·═╗ūāŻ¼╩╣Ųõ”┴-3µ£Īó”┴-4µ£╗“”┴-5µ£ĮY(ji©”)śŗ(g©░u)║═╣”─▄«É│ŻŻ¼▀MČ°ī¦ų┬─IąĪŪ“Īóč█╝░ā╚(n©©i)Č·╗∙Ąū─żĄ─ó¶ą═─zįŁĮY(ji©”)śŗ(g©░u)║═╣”─▄ōp║”Č°ų┬▓ĪĪŻ│ŻęŖĄ─▀zé„ĘĮ╩Įę└┤╬×ķX▀Bµi▀zé„Ż©80ŻźĪ½85ŻźŻ®Īó│Ż╚Š╔½¾wļ[ąįŻ©15ŻźŻ®▀zé„║═│Ż╚Š╔½¾w’@ąį▀zé„Ż©5ŻźŻ®ĪŻ▒Ē¼F(xi©żn)×ķX▀Bµi▀zé„š▀Äū║§Š∙×ķCOL4A5╗∙ę“═╗ūāĪŻįō╗∙ę“╬╗ė┌X╚Š╔½¾wq22Ų¼Č╬Ż¼ŠÄ┤aó¶ą═─zįŁĄ░░ū”┴5µ£Ż©”┴5ĪŠó¶Ī┐Ż®ĪŻ│Ż╚Š╔½¾wļ[ąį▀zé„╝░’@ąį▀zé„š▀×ķCOL4A3╗“COL4A4╗∙ę“═╗ūāŻ¼įō╗∙ę“╬╗ė┌2╠¢╚Š╔½¾wŻ¼ŠÄ┤aó¶ą═─zįŁĄ░░ū”┴3╝░”┴4µ£ĪŻ
═╗ūā╗∙ę“║══╗ūāŅÉą═Ą─▓╗═¼øQČ©Ųõ┼R┤▓▒Ē¼F(xi©żn)Ą─▓Ņ«ÉĪŻCOL4A5╗∙ę“╚▒╩¦╝░ć└ųžĄ─╝¶ĮėÕeš`Ģ■ī¦ų┬ć└ųžĄ──I┼Kōpé¹║═įńŲ┌┬Ā┴”ōp╩¦Ż╗Õe┴x═╗ūā┐╔─▄Ģ■įņ│╔ŪÓ╔┘─Ļą═┬Ā┴”ōp╩¦Īó│╔╚╦ą═░ķ╗“▓╗░ķėą┬Ā┴”ōp╩¦ĪŻCOL4A5╗∙ę“5Ī»─®Č╦╝░ÓÅĮ³COL4A6╗∙ę“5Ī»─®Č╦╚▒╩¦ät┐╔─▄│÷¼F(xi©żn)╩│╣▄║═╔·ų│Ų„ŲĮ╗¼╝Ī┴÷ĪŻCOL4A3╗“COL4A4╗∙ę“Ż©╚Š╔½¾w2Ż®═╗ūāĄ─╝ā║Žūė╗“ļs║ŽūėĢ■▒Ē¼F(xi©żn)×ķ│Ż╚Š╔½¾wļ[ąįĄ─AlportŠC║Žš„Ż¼ļs║Žūė═╗ūāät▒Ē¼F(xi©żn)×ķ┴╝ąį╝ęūÕąįč¬─“Ą─╝▓▓ĪŻ©╚ń╝ęūÕąį▒Ī╗∙Ąū─ż▓ĪTBMDŻ®ĪŻ
┴„ąą▓ĪīWŻ║ė╔ė┌╚▒Ę”┤¾śė▒Š┴„ąą▓ĪīWöĄ(sh©┤)ō■(j©┤)Ż¼░l(f©Ī)▓Ī┬╩╔ą▓╗ŪÕ│■ĪŻ├└ć°ł¾ĖµĄ─AlportŠC║Žš„Ą─╗∙ę“Ņl┬╩×ķ1/10000Ī½1/5000ĪŻ─ąąį║═┼«ąįĄ─░l(f©Ī)▓Ī┬╩╝░▓ĪŪķ▌pųž┼c═╗ūā╗∙ę“ŅÉą═ėąĻP(gu©Īn)ĪŻX▀Bµi▀zé„š▀─ą┼«Š∙┐╔░l(f©Ī)▓ĪŻ¼Ą½─ąąį░l(f©Ī)▓Ī┬╩Ė▀ė┌┼«ąįŻ¼Ūę▓ĪŪķ▌^┼«ąįųžĪŻ
Č■Īó╝▓▓Īį\öÓ
┼R┤▓▒Ē¼F(xi©żn)Ż║AlportŠC║Žš„┐╔ė┌ā║═»Ų┌įńŲ┌Ų▓ĪĪŻĄõą═Ą─┼R┤▓▒Ē¼F(xi©żn)░³└©─I┼KĪóč█▓┐Ė─ūā╝░┬Ā┴”╩▄ōpĪŻ─I┼K▒Ē¼F(xi©żn)×ķč¬─“ĪóĄ░░ū─“║═─I╣”─▄▀MąąąįÉ║╗»Ż¼Ųõųąč¬─“│Ż×ķ│ų└m(x©┤)ąįńRŽ┬č¬─“Ż¼┐╔į┌▀\äė║¾╗“░l(f©Ī)¤ßĢr│÷¼F(xi©żn)╚Ōč█č¬─“Ż¼Ė³│ŻęŖė┌ŪÓ╔┘─Ļą═ĪŻ┬Ā┴”Ė─ūā?y©Łu)ķĖąę¶╔±Į?j©®ng)ąį┬Ā┴”ōp╩¦Ż¼▓óļS▓Ī│╠ųØu╝ėųžŻ¼Ą½ąĶūóęŌ▀Mš╣×ķESRDĄ─X╚Š╔½¾w▀Bµi▀zé„╗╝š▀▓╗ę╗Č©Ģ■│÷¼F(xi©żn)├„’@┬Ā┴”Ž┬ĮĄŻ¼ę“┤╦▓╗─▄īó┬Ā┴”Ž┬ĮĄęĢ×ķAlportŠC║Žš„╣╠ėą╠žš„Ż¼Ę±ät╚▌ęūįņ│╔┬®į\ĪŻč█▓┐«É│Ż▒Ē¼F(xi©żn)×ķĮ³ęĢĪóŪÓ─ĻŁh(hu©ón)║═░ūā╚(n©©i)šŽŻ¼Ą½╚▒Ę”╠ž«ÉąįĪŻ╚²ĘNŠ▀ėąį\öÓęŌ┴xĄ─č█▓┐Ė─ūā░³└©Ż║Ū░łAÕFą╬Š¦ĀŅ¾wĪó║¾ČÓą╬ąįĮŪ─ż╬«┐s║═ęĢŠW(w©Żng)─ż░▀Ż©ęĢŠW(w©Żng)─żųąą─░╝ģ^(q©▒)ė“ų▄ć·Ą─░ū╔½╗“³S╔½Ņw┴ŻŻ®ĪŻ╬ęć°─ąąį╗╝š▀┬Ā┴”╩▄ōp░l(f©Ī)╔·┬╩Ė▀ė┌ć°═Ōł¾Ą└Ż©68Żź▒╚55ŻźŻ®Ż¼┼«ąįätĄ═ė┌ć°═Ōł¾Ą└Ż©7Żź▒╚45ŻźŻ®Ż¼č█«É│Ż░l(f©Ī)╔·┬╩┼cć°═Ōł¾Ą└ŽÓĮ³ĪŻ╔┘öĄ(sh©┤)╗╝š▀▀Ć┐╔│÷¼F(xi©żn)ŲĮ╗¼╝Ī┴÷Ż¼┐╔└█╝░║¶╬³Ą└Īó╬Ė─cĄ└╝░┼«ąį╔·ų│Ą└Ż╗┼╝ėąäė├}┴÷▓ĪūāĪó├µųą▓┐░l(f©Ī)ė²«É│Ż║═Š½╔±░l(f©Ī)ė²▀t£■Ą─ł¾Ą└ĪŻ
╗╝š▀│Żėą╝ęūÕ╩ĘĪŻ│Ż╚Š╔½¾wļ[ąį▀zé„╗╝š▀╗“X▀Bµi▀zé„Ą──ąąį╗╝š▀▓ĪŪķ▀Mš╣▌^┐ņŻ¼│Żė┌16Ī½35Üq▀M╚ļĮK─®Ų┌─I▓ĪŻ╗Č°│Ż╚Š╔½¾w’@ąį▀zé„║═X▀Bµi▀zé„Ą─┼«ąį╗╝š▀ČÓ▓Ī│╠▌^×ķŠÅ┬²Ż¼─I╣”─▄╦źĮ▀│÷¼F(xi©żn)▌^═ĒĪŻį┌═¼ę╗éĆ╗╝▓Ī╝ꎥųąŻ¼╦∙ėą─ąąį│╔åT░l(f©Ī)╔·─I╣”─▄╦źĮ▀Ą──Ļ²gŽÓ╦ŲŻ¼Ą½▓╗═¼╝ꎥųąĄ──ąąį│╔åT░l(f©Ī)╔·─I╣”─▄╦źĮ▀Ą──Ļ²g┐╔ęį▓╗═¼ĪŻę“┤╦Ż¼┐╔Ė∙ō■(j©┤)╗╝▓Ī╝ꎥ░l(f©Ī)╔·─I╣”─▄╦źĮ▀Ą──Ļ²gĘų×ķŻ║ŪÓ╔┘─Ļą═Ż©─I╦źĮ▀░l(f©Ī)╔·į┌ŪÓ╔┘─ĻĢrŲ┌Ż®┼c│╔╚╦ą═Ż©─I╦źĮ▀░l(f©Ī)╔·į┌│╔─Ļęį║¾Ż®ĪŻŲõųąŪÓ╔┘─Ļą═─I═Ō▒Ē¼F(xi©żn)Ė³×ķ═╗│÷Ż¼─ą║ó═©│Żį┌Ę▒č▄║¾┤·Ū░╝┤╦└═÷Ż¼ų┬▓Ī╗∙ę“│Ż×ķą┬░l(f©Ī)═╗ūāŻ╗Č°│╔╚╦ą═╝ꎥųąĄ─╗╝š▀▌^─ĻķLŻ¼▌^╔┘ą┬░l(f©Ī)═╗ūāĪŻ
▌oų·Öz▓ķŻ║
1.īŹ“×╩ęÖz▓ķ
─“│ŻęÄ(gu©®)Öz▓ķ’@╩ŠńRŽ┬č¬─“║═Ą░░ū─“ĪŻ─I╣”─▄Öz▓ķ╠ß╩Šč¬╝Ī¶¹ųØu╔²Ė▀ūŅĮK▀_ĄĮĮK─®Ų┌─I▓Ī╦«ŲĮĪŻļS─I╣”─▄É║╗»Ż¼▀ĆĢ■░ķ░l(f©Ī)Ųõ╦¹╗»“׫É│ŻĪŻ╚ńč¬│ŻęÄ(gu©®)Öz▓ķ╠ß╩Šš²╝Ü░¹š²╔½╦žąįžÜ謯¼┤·ųxąį╦ßųąČŠ╝░ļŖĮŌ┘|(zh©¼)«É│ŻŻ¼Ą═č¬Ō}ĪóĖ▀č¬┴ūĪóč¬╝ūĀŅ┼įŽ┘╦ž╦«ŲĮ╔²Ė▀Ą╚ĪŻ
2.ļŖ£y┬Ā
įńŲ┌▒Ē¼F(xi©żn)×ķĖ▀ŅlĘČć·┬Ā┴”Ž┬ĮĄĪŻļS▓Ī│╠▀Mš╣Ż¼┬Ā┴”Ž┬ĮĄĘČć·ųØuöU┤¾Ż¼╔§ų┴░l(f©Ī)š╣×ķ╚½ę¶ė“ĪŻļpé╚(c©©)Č·├@│╠Č╚┐╔▓╗═Ļ╚½ī”ĘQĪŻ
3.č█┐ŲÖz▓ķ
┐╔░l(f©Ī)¼F(xi©żn)ęĢ┴”Ž┬ĮĄĪó░ūā╚(n©©i)šŽŻ¼Ą½Š▀ėąį\öÓąįĄ─╚²ŅÉ▓Īūā?y©Łu)ķŻ║Ū░łAÕFą╬Š¦ĀŅ¾wĪó║¾ČÓą╬ąįĮŪ─ż╬«┐s║═ęĢŠW(w©Żng)─ż░▀ĪŻ
4.ĮM┐Ś▓Ī└ĒĖ─ūā
Ż©1Ż®─I┼K╗ŅÖz
╠žš„ąįĖ─ūāąĶļŖūė’@╬óńRŽ┬ė^▓ņĄĮ─IąĪŪ“╗∙Ąū─żŻ©GBMŻ®ÅVĘ║į÷║±Īóūā▒Īęį╝░ų┬├▄īėŠW(w©Żng)╗@ĀŅ┐v┴čĘųīėĪŻ╣ŌńRŽ┬¤o╠žš„ąįĄ─▓Ī└Ēūā╗»Ż¼Ą½│Ż┐╔ęŖĄĮGBM╚Š╔½▓╗┴╝Īóŗļā║śė─IąĪŪ“║═─Iķg┘|(zh©¼)┼▌─Ł╝Ü░¹Ż¼─IąĪŪ“┐╔│÷¼F(xi©żn)ŠųįŅ╣Ø(ji©”)Č╬ŽĄ─żōpé¹║═├½╝Üč¬╣▄▒┌į÷║±Ż¼╝s30Żź─IąĪŪ“┐╔ęŖŪ“─ęš│▀BĪŻ├Ōę▀¤╔╣ŌīWÖz▓ķę▓¤o╠ž«ÉąįŻ¼ėąĢr┐╔ęŖŽĄ─żģ^(q©▒)╝░čžGBM╣Ø(ji©”)Č╬╗“Åø┬■ąįŅw┴ŻĀŅC3║═IgM│┴ĘeŻ¼ėąĢr×ķ╚½ĻÄąįĪŻ
Ż©2Ż®Ųż─w╗ŅÖz
╣ŌńRø]ėą╠ž«ÉąįĖ─ūāĪŻĄ½Ųż─w║═─I┼KĮM┐ŚĄ─ó¶ą═─zįŁ├Ōę▀╚Š╔½┐╔░l(f©Ī)¼F(xi©żn)ó¶ą═─zįŁ”┴-3µ£Īó”┴-4µ£║═Ż©╗“Ż®”┴-5µ£╚▒╩¦╗“«É│ŻĘų▓╝ĪŻ
5.╗∙ę“Öz▓ķ
╩Ūį\öÓ▒Š▓ĪĄ─Įś╦£╩ĪŻ┐╔▒Ē¼F(xi©żn)×ķCOL4A3ĪóCOL4A4╗“COL4A5╗∙ę“╚▒Ž▌ĪŻ
į\öÓŻ║AlportŠC║Žš„į\öÓę└┐┐┼R┤▓▒Ē¼F(xi©żn)ĪóĮM┐Ś▓Ī└ĒĪó╝ꎥĘų╬÷╝░╗∙ę“į\öÓĪŻ┬²ąį─IčūŠC║Žš„║Ž▓ó┬²ąį─I╣”─▄▓╗╚½Ą─╗╝š▀Ż¼╚ń╣¹ėą╝ęūÕ╩ĘŻ¼═¼Ģr┤µį┌Ė▀ŅlĘČć·┬Ā┴”Ž┬ĮĄĪóŪ░łAÕFą╬Š¦ĀŅ¾wĪó╗“║¾ČÓą╬ąįĮŪ─ż╬«┐sĪó╗“ęĢŠW(w©Żng)─ż░▀Ż╗▓Ī└ĒÖz▓ķ░l(f©Ī)¼F(xi©żn)─I┼KGBM│÷¼F(xi©żn)ÅVĘ║Ą─į÷║±Īóūā▒Īęį╝░ų┬├▄īė┐v┴čĘųīėĄ─╠žš„ąįĖ─ūāŻ¼─I┼K╗“Ųż─wĮM┐Śó¶ą═─zįŁ├Ōę▀╚Š╔½░l(f©Ī)¼F(xi©żn)ó¶ą═─zįŁ”┴-3µ£Īó”┴-4µ£║═Ż©╗“Ż®”┴-5µ£╚▒╩¦╗“«É│ŻĘų▓╝Ż¼ätĖ▀Č╚╠ß╩Š▒Š▓ĪŻ¼┤_į\ąĶę¬Öz£yCOL4A3ĪóCOL4A4╗“COL4A5╗∙ę“╚▒Ž▌ĪŻ
Ķbäeį\öÓŻ║
1.ąĶę¬┼c▒Ē¼F(xi©żn)×ķč¬─“ĪóĄ░░ū─“╝░─I╣”─▄ōp║”Ą─┬²ąį─IąĪŪ“─IčūŻ¼ė╚Ųõ╩ŪIgA─I▓Ī║═▒Ī╗∙Ąū─ż─I▓ĪĶbäeĪŻ┐╔═©▀^╝ęūÕ╩ĘĪó┬Ā┴”Öz£yĪóč█┐ŲÖz▓ķĪóŲż─w╗“─I┼K╗ŅÖz▀MąąĶbäeĪŻ
2.╚ń═¼Ģr┤µį┌─IčūĪóĖąę¶ąįČ·├@▓Ī╩Ę╝░╝ęūÕ╩Ęš▀Ż¼▀ĆąĶ┼cEpsteinŠC║Žš„/FechtnerŠC║Žš„▀MąąĶbäeŻ¼▀@ā╔ŅÉ╝▓▓Ī×ķ22╠¢╚Š╔½¾wŠÄ┤aĘŪ╝Ī╚Ō╝ĪŪ“Ą░░ūųžµ£9Ż©MYH9Ż®Ą─╗∙ę“═╗ūā╦∙ų┬Ż¼┐╔═©▀^╗∙ę“į\öÓĶbäeĪŻ
3.╦Ä╬’╚ń░▒╗∙╠Ū▄šŅÉ┐╣╔·╦ž┐╔═¼Ģrįņ│╔┬Ā┴”Ž┬ĮĄ╝░─I╣”─▄«É│ŻŻ¼ąĶūą╝Üįāå¢▓Ī╩ĘĪóė├╦Ä╩ĘÄ═ų·ĶbäeĪŻ
╚²Īóų╬»¤ĘĮ╩Į
ų╬»¤Ż║AlportŠC║Žš„Ģ║¤oĖ∙ų╬»¤Ę©ĪŻ─┐Ū░ų╬»¤ęįų¦│ų║═─I┼K╠µ┤·ų╬»¤×ķų„ĪŻ
1.ĘŪ╠ž«Éąį╦Ä╬’Ė╔ŅA
─I╦ž-č¬╣▄ŠoÅł╦žŽĄĮy(t©»ng)Ż©RAASŻ®ęųųŲ䮯¼░³└©č¬╣▄ŠoÅł╦ž▐D(zhu©Żn)╗»├ĖęųųŲä®Īóč¬╣▄ŠoÅł╦žó“╩▄¾w▐ū┐╣ä®║═╚®╣╠═¬╩▄¾w▐ū┐╣䮥╚┐╔ęį═©▀^ęųųŲRAAS╗Ņ╗»Īóš{(di©żo)š¹Ū“╣▄Ę┤üŻ¼ĮĄĄ═─IąĪŪ“Ė▀×V▀^Č°£p╔┘Ą░░ū─“Ż¼čėŠÅ─IąĪŪ“ė▓╗»║═╝▓▓Ī▀Mš╣ĪŻĄ½ąĶę¬▒O(ji©Īn)£yč¬Ōø║═─I╣”─▄Ż¼Š»╠ĶĖ▀Ōøč¬░Y║═─I╣”─▄┐ņ╦┘▀Mš╣Ą─Ė▒ū„ė├ĪŻ┤╦═ŌŻ¼ąĶī”░Y╠Ä└Ē─I╣”─▄▓╗╚½╦∙ĦüĒĄ─▓ó░l(f©Ī)░YŻ¼╚ń─IąįĖ▀č¬ē║ĪóŌ}┴ū┤·ųx«É│ŻĪó─IąįžÜč¬║═ļŖĮŌ┘|(zh©¼)Īó╦ßēAŲĮ║Ō╬╔üyĪŻ
2.─I┼K╠µ┤·ų╬»¤
ī”ė┌▀Mš╣ĄĮĮK─®Ų┌─I▓ĪĄ─╗╝š▀ąĶ▀Mąą─I┼K╠µ┤·ų╬»¤Ż¼┐╔▀xō±č¬ę║═Ė╬÷ĪóĖ╣─ż═Ė╬÷║═─I┼KęŲų▓ĪŻ─IęŲų▓ąg(sh©┤)║¾ąĶŠ»╠Ķ░l(f©Ī)╔·┐╣─IąĪŪ“╗∙Ąū─ż▓ĪŻ¼ė╚Ųõ╩ŪCOL4A5╗∙ę“═╗ūā╗╝š▀Ė³ąĶĻP(gu©Īn)ūóĪŻ
3.Č·▒Ū║Ē┐Ų╠Ä└Ē
ų·┬ĀŲ„ėąų·ė┌Ė─╔ŲŽ┬ĮĄĄ─┬Ā┴”Ż¼Ą½▓╗─▄═Ļ╚½╝mš²┬Ā┴”«É│ŻŻ╗Č·°Q═©│Żī”╚╬║╬ų╬»¤¤oą¦Ż¼ų·┬ĀŲ„┐╔═©▀^öU┤¾═Ōų▄┬Ģę¶Č°£pąĪČ·°QĖ╔ö_ĪŻ
4.č█┐Ų╠Ä└Ē
ęĢŠW(w©Żng)─ż▓Īūā═©│Ż▓╗Ģ■ė░ĒæęĢ┴”Ż¼▓╗ąĶę¬ų╬»¤Ż╗łAÕFŠ¦ĀŅ¾w╗“░ūā╚(n©©i)šŽįņ│╔Ą─ć└ųžęĢ┴”ōp║”▓╗─▄═©▀^č█ńR╗“ļ[ą╬č█ńR│Cš²Ż╗Š¦ĀŅ¾wš¬│²╝░č█ā╚(n©©i)Š¦¾wų▓╚ļ╩Ūąąų«ėąą¦Ą─ś╦£╩ų╬»¤ĪŻ
5.╗╝š▀╣▄└Ē
░³└©╝▓▓ĪČÓīW┐ŲŠC║Ž╣▄└ĒĪŻĮ©ūhį┌─I┼Kā╚(n©©i)┐ŲĪóČ·▒Ū║Ē┐Ų╝░č█┐ŲęÄ(gu©®)┬╔ļSį\Ż¼Č©Ų┌įuār╗╝š▀┬²ąį─I▓Ī▓ó░l(f©Ī)░YĪó┬Ā┴”╝░ęĢ┴”Ė─ūāĪŻ▒▄├Ō─IČŠąį╦Ä╬’ĪóČ·ČŠąį╦Ä╬’Ż¼▒▄├ŌķLŲ┌▒®┬Čė┌Ė▀įļ궣h(hu©ón)Š│ĪŻ
6.▀zé„ū╔įā
╗╝š▀╝░╗∙ę“öyĦš▀ąĶę¬▀Mąą▀zé„ū╔įāŻ¼▒žę¬ĢrąĶę¬▀Mąą«a(ch©Żn)Ū░į\öÓĪŻ
į\»¤┴„│╠łDŻ║
ģó┐╝╬─½IŻ║
[1] ═§║ŻčÓ.─I┼K▓ĪīW.Ą┌2░µ.▒▒Š®:╚╦├±ąl(w©©i)╔·│÷░µ╔ń,2012Ż║1777.
[2] Maarten W. Toal, Glenn M. Chertow, Philip A.Marsden, et al. Brenner and RectorĪ»s The Kidney. 9th Edition. Philadelphia. Saunders , 2011:1237
[3] Clifford E Kashtan, MD. Clinical manifestations, diagnosis, and treatment of Alport syndrome (hereditary nephritis). Up To Date. Up To Date Inc, 2018.
[4] Savige J, Gregory M, Gross O, et al. Expert guidelines for the management of Alport syndrome and thin basement membrane nephropathy. J Am Soc Nephrol,2013, 24(3):364-375.
[5] ═§Ę╝,ČĪØŹ,ėßČYŽ╝,Ą╚.ųąć°AlportŠC║Žš„┼R┤▓╠žš„.ųąć°ā║┐ŲļsųŠ,2003,21(10):601-604.
[6] Zhang Y, Ding J. Renal, auricular, and ocular outcomes of Alport syndrome and their current management. Pediatr Nephrol,2018,33(8):1309-1316.
░µÖÓ(qu©ón)┬Ģ├„
ęį╔Žā╚(n©©i)╚▌üĒūį┴╝ßt(y©®)ģR-║▒ęŖ▓Īą┬▀Mš╣Ż¼╚ńėąĮ©ūh╗“ę╔å¢Ż¼ÜgėŁų┬ļŖ18017449015ĪŻ
 ķL░┤Č■ŠS┤a
ķL░┤Č■ŠS┤aĻP(gu©Īn)ūóŠ½▓╩ā╚(n©©i)╚▌





