
ę╗Īó╗∙▒Šą┼Žó
Ė┼╩÷Ż║ļĶ╣Ū╝Ī╬«┐s░YŻ©Charcot-Marie-Tooth diseaseŻ¼CMTŻ®╩Ūę╗ĮM▀zé„ąįų▄ć·╔±Įø▓ĪĪŻ─┐Ū░ęč░l¼FĄ─ų┬▓Ī╗∙ę“▀_60ėÓĘNĪŻŲõų„ę¬╠ž³c×ķ┬²ąį▀MąąąįĪóķLČ╚ę└┘ćĄ─▀\äė╝░ĖąėX╔±Įø▓ĪŻ¼ūŅ│ŻęŖ▒Ē¼F×ķŽ┬ų½Ų▓ĪĄ─ĪóŠÅ┬²▀Mš╣Ą─ų½¾w▀hČ╦╝Ī╚Ō╬«┐sŻ¼¤o┴”║═ĖąėX╚▒╩¦ĪŻĖ∙ō■╔Žų½▀\äė╔±Įøé„ī¦╦┘Č╚ų„ę¬Ęų×ķ╦ĶŪ╩ą═║═▌S╦„ą═ĪŻĖ∙ō■▀zé„ĘĮ╩ĮĪó┼R┤▓▒Ē¼Fęį╝░ļŖ╔·└ĒŻ¼CMTų„ę¬üåą═░³└©CMT1-4ęį╝░CMTXĪŻ┤╦═Ō▀ĆėąCMT5-7ĪódHMNŻ©▀hČ╦ą═▀zé„ąį▀\äė╔±Įø▓ĪŻ®ĪóHNPPŻ©▀zé„ē║Ų╚ęūĖąų▄ć·╔±Įø▓ĪŻ®ĪŻį┌├┐éĆüåą═ųąŻ¼▓╗═¼ūų─Ė┤·▒Ē▓╗═¼╗∙ę“═╗ūāŻ©╚ńCMT1AŻ¼CMT1BŻ®ĪŻ
▓Īę“Ż║CMT×ķę╗ĮMė╔▓╗═¼╗∙ę“═╗ūāī¦ų┬Ą─ų▄ć·╔±Įø▓ĪĪŻ▀@ą®╗∙ę“ŠÄ┤aĄ─Ą░░ū▒Ē▀_ė┌ų▄ć·╔±ĮøĄ─╦ĶŪ╩╗“▌S╦„Ż¼═╗ūāī¦ų┬ų▄ć·╔±Įø╦ĶŪ╩ą╬│╔╚▒Ž▌╗“▌S╦„╣”─▄«É│ŻĪŻ
▀zé„ĘĮ╩ĮĘų×ķ│Ż╚Š╔½¾w’@ąį▀zé„Īó│Ż╚Š╔½¾wļ[ąį▀zé„║═X▀Bµiļ[ąį▀zé„Ą╚ĪŻ│ŻęŖĄ─═╗ūā╗∙ę“░³└©PMP22Ż¼MPZŻ¼GJB1Ż¼MFN2ĪŻ│ŻęŖĄ─üåą═×ķCMT1Ż¼CMT2Ż¼CMTXĪŻCMT1×ķ│Ż╚Š╔½¾w’@ąį▀zé„Ą─├ō╦ĶŪ╩ąįCMTŻ¼ŲõųąCMT1A×ķūŅ│ŻęŖĄ─CMTüåą═Ż©š╝40Ī½50ŻźŻ®Ż¼Ųõ═╗ūā╗∙ę“×ķPMP22Ż╗CMT1Bš╝CMT1Ą─3Ī½5ŻźŻ¼Ųõ═╗ūā╗∙ę“×ķMPZĪŻX▀Bµiļ[ąį▀zé„Ą─CMTX1×ķĄ┌2│ŻęŖĄ─CMTüåą═Ż©š╝10ŻźŻ®Ż¼Ųõ═╗ūā╗∙ę“×ķGJB1ĪŻCMT2×ķ▌S╦„ąįCMTŻ¼Ųõųą│ŻęŖĄ─═╗ūā╗∙ę“░³└©MFN2Ż©š╝CMT2Ą─20ŻźŻ®ĪóMPZŻ©š╝CMT2Ą─5ŻźŻ®ĪóNEFL║═GDAP1Ą╚ĪŻCMT4×ķ│Ż╚Š╔½¾wļ[ąį▀zé„Ą─├ō╦ĶŪ╩ąįCMTŻ¼ŲõūŅ│ŻęŖĄ─═╗ūā╗∙ę“×ķGDAP1ĪŻPMP22ųžÅ═ĪóGJB1═╗ūāĪóPMP22╚▒╩¦ĪóMPZ═╗ūāĪóMFN2═╗ūā▀@5ĘNüåą═š╝╦∙ėąCMTĄ─92ŻźĪŻ
┴„ąą▓ĪīWŻ║CMTĄ─┐é¾w░l▓Ī┬╩╝s×ķ40/100 000Ż¼░l▓Ī┬╩į┌╚╦ĘNķg¤o├„’@▓ŅäeĪŻ
Č■Īó╝▓▓Īį\öÓ
┼R┤▓▒Ē¼FŻ║CMTĄ─ų„ę¬┼R┤▓▒Ē¼F×ķŽ┬ų½▀hČ╦×ķų„Ż¼▓óųØuŽ“Į³Č╦░lš╣Ą─ų½¾w╝Ī╚Ō╬«┐sĪó¤o┴”╝░ĖąėXå╩╩¦ĪŻ│ŻęŖ┼R┤▓▒Ē¼F×ķ▀\äė─▄┴”▓╗╚ń═¼²g╚╦Ż¼┼▄▓Į└¦ļyŻ¼ęū┼ż─_Ż¼ūŃŽ┬┤╣Ż¼ąĪ═╚ļĶ─c╝Ī╬«┐są╬╦ŲĪ░·Q═╚Ī▒Ż╗▓ķ¾w┐╔░l¼F╣Łą╬ūŃĪóÕNĀŅų║Ż¼▀hČ╦ų½¾w×ķų„Ą─¤o┴”╬«┐sŻ¼╔ŅĖąėX£p═╦ĪŻ╗╝š▀═©│Ż20ÜqŪ░Ų▓ĪŻ¼ŠÅ┬²▀Mš╣Ż¼╝▓▓Ī║¾Ų┌┐╔─▄ć└ųžė░Ēæ╗ŅäėŻ¼Ą½║▄╔┘ī¦ų┬═Ļ╚½Üł╝▓Ż¼ę▓▓╗ė░Ēæš²│Żē█├³ĪŻĄ½ėąą®╠ž╩ŌŅÉą═┐╔─▄Ų▓ĪįńŪęć└ųžŻ║╚ńDejerine-SottasŠC║Žš„╗╝š▀ŗļā║Ų┌Ų▓ĪŻ¼ī¦ų┬Ą═Åł┴”Ą─▄øŗļĪó▀\äė░lė²▀t£■Ą╚ĪŻ╔┘öĄCMT┐╔ėąų▄ć·╔±Įø▓Īęį═ŌĄ─Ųõ╦¹▒Ē¼FŻ║CMTX1ą═┐╔ėąūõųąśė░lū„░ķMRI░ū┘|┐╔─µąį▓ĪūāŻ╗CMT5ą═░ķÕF¾w╩°š„Ż╗CMT6ą═░ķęĢ╔±Įø╬«┐sŻ╗CMT7ą═░ķ╔½╦žąįęĢŠW─żčūĪŻ
▌oų·Öz▓ķŻ║
1.ļŖ╔·└ĒÖz▓ķ
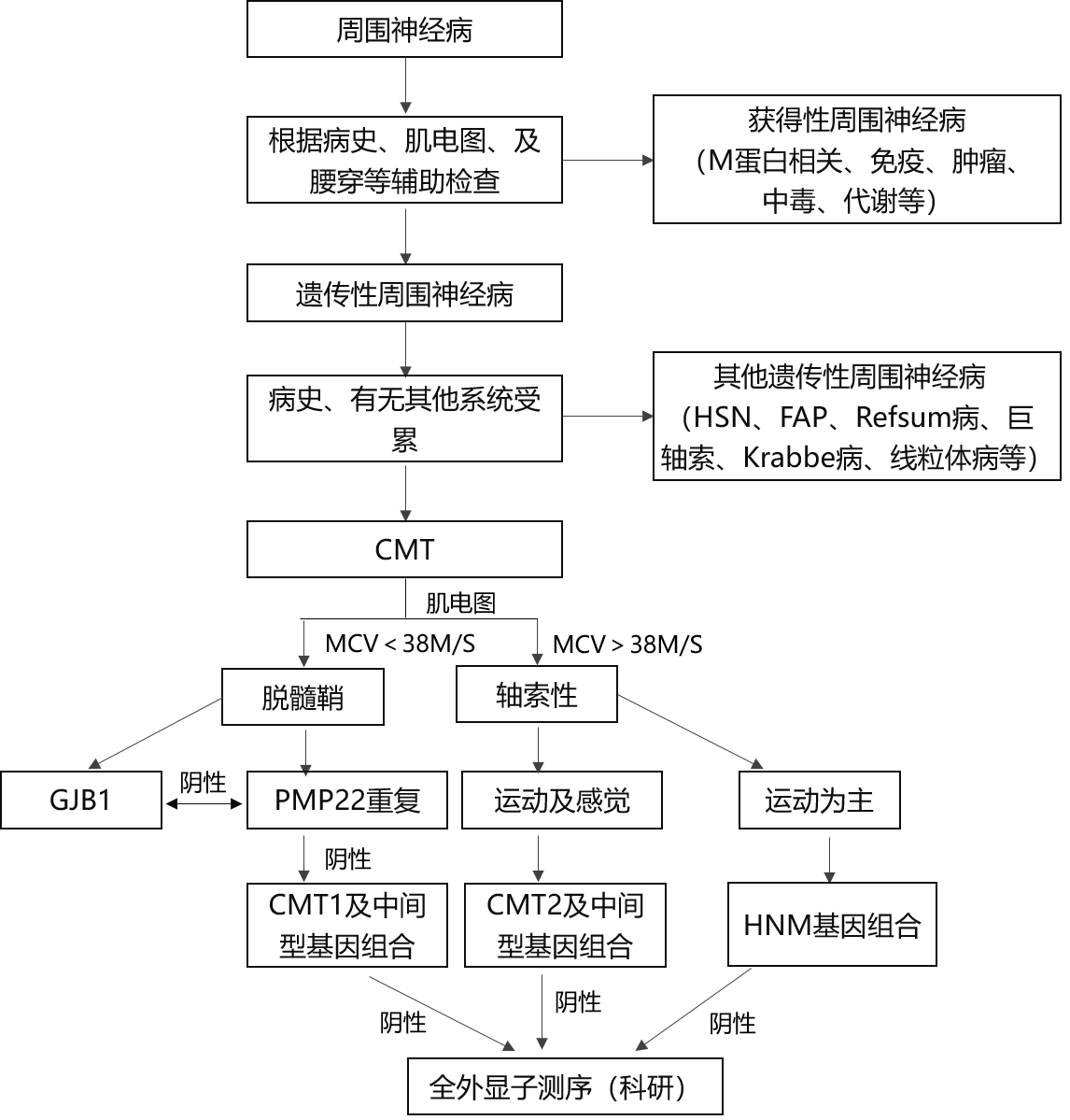
ļŖ╔·└ĒÖz▓ķī”ė┌ģ^Ęų├ō╦ĶŪ╩ąį║═▌S╦„ąį╔±Įø▓Ī╩«Ęųųžę¬Ż¼═¼Ģr┐╔ęįÖz£y╩Ūʱėą┼R┤▓Ž┬Ą─ĖąėX╔±Įø╩▄└█Ż¼ėąų·ė┌CMTĄ─Ęųą═Ż╗┤╦═ŌŻ¼╣ØČ╬ąį▀\äė╔±Įøé„ī¦Öz£yį┌├ō╦ĶŪ╩ą═CMT┼cCIDPĄ─Ķbäeųąę▓ėąųžę¬ū„ė├ĪŻŠ∙ä“Ą─╔±Įøé„ī¦╦┘Č╚£p┬²Ż©╔Žų½▀\äė╔±Įøé„ī¦╦┘Č╚Ż╝38m/sŻ®╠ß╩Š├ō╦ĶŪ╩ą═CMTŻ©CMT1ęį╝░CMT4Ż®Ż¼Č°╔±Įøé„ī¦╦┘Č╚š²│Ż╗“▌pČ╚£p┬²Ż©š²ųą╗“│▀╔±Įø▀\äėé„ī¦╦┘Č╚ŻŠ38m/sŻ®Īó░ķėąÅ═║Ž╝Ī╚Ōäėū„ļŖ╬╗╝░ĖąėXäėū„ļŖ╬╗▓©Ę∙ĮĄĄ═╠ß╩ŠCMT2ĪŻ«ö╔Žų½Ą─▀\äė╔±Įøé„ī¦╦┘Č╚╬╗ė┌25Ī½45m/sĄ─ųąķgųĄĢrŻ¼ąĶꬊ»╠ĶCMTX1ĪŻ├ō╦ĶŪ╩ą═CMTĄ─▀\äė╔±Įøé„ī¦╦┘Č╚═©│ŻŠ∙ä“£p┬²Ż¼╚¶│÷¼F├„’@Ą─▓©ą╬ļx╔óĪóé„ī¦ūĶ£■═©│Ż╠ß╩ŠCIDP┐╔─▄ąį┤¾ĪŻĄ½į┌MPZ╗∙ę“═╗ūāĄ─CMT1BųąŻ¼┼╝Ā¢Ģ■│÷¼Fé„ī¦ūĶ£■Ż╗CMTX1ųąŻ¼ėąĢr┐╔ėą▓╗ī”ĘQĄ─é„ī¦╦┘Č╚£p┬²Ż¼┐╔ėą├„’@Ą─▓©ą╬ļx╔ó╔§ų┴é„ī¦ūĶ£■ĪŻ
2.▀zé„īWÖz▓ķ
╗∙ę“Öz£yī”ė┌CMTĄ─į\öÓ║═Ęųą═╩«Ęųųžę¬ĪŻĶbė┌PMP22ĪóMPZĪóGJB1╝░MFN2╗∙ę“═╗ūāį┌CMTųąš╝90Żźęį╔ŽŻ¼╣╩┐╔ęįĖ∙ō■╗╝š▀Ą─┼R┤▓║═ļŖ╔·└Ē╠žš„▀xō±┐╔─▄ŽÓĻPĄ─╗∙ę“▀Mąąę╗┤·£yą“Öz£yĪŻļSų°Ė▀═©┴┐£yą“╝╝ągĄ─Ųš╝░Ż¼ī”ė┌│Ż╚Š╔½¾w’@ąįĄ─├ō╦ĶŪ╩ąįų▄ć·╔±Įø▓ĪŻ¼┐╔╩ūŽ╚▓╔ė├MLPA╝╝ąg▀MąąPMP22╗∙ę“ųžÅ══╗ūāĄ─Öz£yŻ¼╚ń╣¹ĻÄąįį┘▀xō±Ė▀═©┴┐£yą“ĘĮĘ©ī”Ė³ČÓŽÓĻP╗∙ę“▀MąąÖz£yĪŻ
3.╔±Įø▓Ī└ĒÖz▓ķ
ļSų°╗∙ę“Öz£yĘĮĘ©æ¬ė├Ż¼Į^┤¾ČÓöĄę╔į\▓Ī└²¤oąĶ▀Mąą╔±Įø╗ŅÖzĪŻĄ½«ö┼R┤▓╝░╝ĪļŖłD▓╗Ąõą═ĢrŻ¼┐╔═©▀^╔±Įø╗ŅÖzüĒģfų·Ķbäeį\öÓĪŻ
į\öÓŻ║CMTĄ─į\öÓę└┐┐┼R┤▓▒Ē¼F║═¾wĖ±Öz▓ķĪóļŖ╔·└ĒÖz▓ķ╝░╗∙ę“Öz£yĪŻī”ė┌ŠÅ┬²▀Mš╣Ą─ų½¾w▀hČ╦╝Ī╚Ō¤o┴”╬«┐sĪó╣Łą╬ūŃĪó░ķ╗“▓╗░ķėą▌pČ╚ĖąėX«É│ŻŻ¼ļŖ╔·└Ē╠ß╩ŠĖąėX▀\äėąįų▄ć·╔±Įø▓ĪĄ─╗╝š▀Ż¼¤ošōėą¤oĻ¢ąį╝ęūÕ╩ĘŻ¼ąĶ┐╝æ]ĄĮ▀zé„ąįų▄ć·╔±Įø▓ĪŻ¼╠žäe╩ŪCMTĪŻ╗∙ę“Öz£y╩Ū┤_į\CMT╝░▀MąąĘųą═Ą─║╦ą─╩ųČ╬ĪŻ
«aŪ░į\öÓŻ║ī”ė┌ć└ųžų┬ÜłĄ─ŅÉą═Ż¼į┌╝ęī┘│õĘųų¬ŪķĪóš„Ū¾ęŌęŖ║¾Ż¼┐╔┐╝æ]į┘┤╬╔·ė²Ģr▀Mąą«aŪ░į\öÓĪŻ
Ķbäeį\öÓŻ║
1.CMTų„ꬹĶę¬┼cę╗ą®└█╝░ų▄ć·╔±ĮøĄ─Ųõ╦¹▀zé„ąį╝▓▓ĪŽÓĶbäeŻ¼╚ńKrabbe─X░ū┘|ĀIB▓╗┴╝Īó«É╚Šąį─X░ū┘|ĀIB▓╗┴╝ĪóŠĆ┴Ż¾w▓ĪĪó▀zé„ąį»döüąįĮž░c║═▀zé„ąį╣▓Ø·╩¦š{Ą╚ĪŻ╦³éā│²Š▀ėąų▄ć·╔±Įø▓Īęį═ŌŻ¼▀Ćėą╔±ĮøŽĄĮyŲõ╦¹▓┐╬╗║═ĘŪ╔±ĮøĮM┐ŚŲ„╣┘╩▄└█Ą─▒Ē¼FŻ¼Č°CMT▌^╔┘ėąų▄ć·╔±Įøęį═ŌĄ─Ųõ╦¹ŽĄĮy╩▄└█ĪŻ┴Ēę╗ą®ęįų▄ć·╔±Įø╩▄└█×ķų„Ą─▀zé„ąį▓ĪŻ¼╚ń▀hČ╦▀zé„ąį▀\äė╔±Įø▓ĪŻ©dHMNŻ®ĪóRefsum▓ĪĪó╝ęūÕąįĄĒĘ█śėūāąįĪóŠ▐▌S╦„╔±Įø▓Ī║═▀zé„ąįē║Ų╚ęūĖąų▄ć·╔±Įø▓ĪĄ╚Ż¼ąĶę¬į┌┼R┤▓║═ļŖ╔·└ĒÖz▓ķ╗∙ĄA╔ŽŻ¼▀xō±▒žę¬Ą─╔·╗»Öz“×Īó╔±Įø╗ŅÖz▓Ī└Ē║═╗∙ę“Öz£yüĒ╝ėęįĶbäeĪŻ┤╦═ŌŻ¼▀ĆąĶę¬┼c▀hČ╦ą═╝Ī▓Ī║═Ž┬▀\äė╔±Įøį¬ŠC║Žš„Ż©╚ń╝╣╝Ī╬«┐s░YŻ®ŽÓĶbäeŻ¼╝ĪļŖłD╝░▒žę¬Ą─╝Ī╚Ō╗ŅÖz▓Ī└Ē╝░╗∙ę“Öz£yėąų·ė┌Ķbäeį\öÓĪŻ
2.CMTąĶę¬┼c½@Ą├ąįų▄ć·╔±Įø▓ĪŽÓĶbäeŻ¼╚ńCIDPŻ©┬²ąįčūąį├ō╦ĶŪ╩ąįČÓ░l╔±ĮøĖ∙ų▄ć·╔±Įø▓ĪŻ®ĪóĖ▒Ą░░ūč¬░YŽÓĻPų▄ć·╔±Įø▓ĪŻ¼▌S╦„ąį╚ńųąČŠĪó┤·ųxŽÓĻPų▄ć·╔±Įø▓Ī║═ČÓįŅ▀\äė╔±Įø▓ĪĄ╚ĪŻCMT═©│Żį┌ŪÓ╔┘─Ļ╗“ėū─ĻŲ▓ĪŻ¼Ų▓Ī─Ļ²g═ĒąĶŠ»╠Ķ½@Ą├ąįų▄ć·╔±Įø▓ĪĪŻCMTČÓŲ▓Īļ[─õĪóöĄ─Ļā╚ŠÅ┬²╝ėųžŻ¼Č°½@Ą├ąįų▄ć·╔±Įø▓ĪČÓ▓Ī│╠▌^Č╠ĪŻ▓ķ¾w░l¼F╣Łą╬ūŃĪóÕNĀŅų║Īó·Q═╚░YĀŅ╠ß╩ŠCMT┐╔─▄ąį┤¾ĪŻ├ō╦ĶŪ╩ą═CMTĄ─▀\äė╔±Įøé„ī¦╦┘Č╚═©│ŻŠ∙ä“£p┬²Ż¼╚¶│÷¼F├„’@Ą─▓©ą╬ļx╔óĪóé„ī¦ūĶ£■Ż¼═©│Ż╠ß╩ŠCIDP┐╔─▄ąį┤¾ĪŻCMTĄ──X╝╣ę║Ą░░ū┐╔▌pČ╚╔²Ė▀Ż¼Ą½╚¶├„’@╔²Ė▀Ż©╚ńŻŠ1g/LŻ®ätąĶ┐╝æ]CIDPĄ╚½@Ą├ąįų▄ć·╔±Įø▓Ī┐╔─▄ĪŻ
╚²Īóų╬»¤ĘĮ╩Į
ų╬»¤Ż║─┐Ū░Ż¼CMTĄ─ų╬»¤ų„ę¬╩Ūų¦│ųų╬»¤Ż¼ø]ėąĖ─╔Ų╝▓▓ĪĄ─╠ž«Éąį╦Ä╬’ĪŻ▀m«öĄ─ų¦│ųų╬»¤─▄ē“’@ų°Ė─╔Ų╗╝š▀Ą─╔·╗Ņ┘|┴┐ĪŻ
1.┐ĄÅ═ų╬»¤
ęÄĘČĄ─┐ĄÅ═ų╬»¤─▄ē“čėŠÅ╝▓▓Īįņ│╔Ą─╣”─▄šŽĄK╚ńĻP╣Ø╗¹ą╬Ą╚Ż¼ŠS│ųĖ³║├Ą─╔·╗Ņ╣”─▄║═ū╦æBĪŻų¦Š▀ą¼Ą╚┐╔Ė─╔Ųąąū▀▓ĮæBĪŻ
2.═Ō┐Ų│Cą╬ų╬»¤
ī”ė┌ć└ųžĄ─╣Ū„└╗¹ą╬Ż¼╠žäe╚ńĖ▀ūŃ╣ŁĪóÕNĀŅų║╗¹ą╬Ż¼╩ųąg│Cą╬┐╔─▄ėąęµĪŻ
3.▒M┴┐▒▄├Ō╩╣ė├┐╔─▄╝ėųžCMTĄ─╦Ä╬’
╚ńķL┤║ą┬ēAĪó░ĘĄŌ═¬Īó┼╠µū¶├ūĪóŃKŅÉĪó░▒▒ĮĒ┐ĪóüĒĘ·├ū╠žĪó▀╗Ó½═ūę“Īó╝ūŽ§▀“Īó╦Š╦¹Ę“Č©Īó╦¹┐╦─¬╦ŠĪó╔│└¹Č╚░ĘĪóį·╬„╦¹×IĄ╚ĪŻ
4.▀zé„ū╔įā┼c«aŪ░į\öÓ
CMTŅÉą═▒ŖČÓŻ¼╗∙ę“┤_į\║¾Į©ūh▀zé„ū╔įāŻ¼├„┤_▓Īę“╝░╝ꎥ│╔åT’LļUĪŻī”ė┌ć└ųžų┬ÜłĄ─ŅÉą═Ż¼į┌╝ęī┘│õĘųų¬ŪķĪóš„Ū¾ęŌęŖ║¾Ż¼┐╔┐╝æ]į┘┤╬╔·ė²Ģr▀Mąą«aŪ░į\öÓĪŻ
į\»¤┴„│╠łDŻ║
ļĶ╣Ū╝Ī╬«┐s░YŻ©CMTŻ®į\»¤┴„│╠
▀zé„ū╔įāŻ║CMTŅÉą═▒ŖČÓŻ¼╗∙ę“┤_į\║¾Į©ūh▀zé„ū╔įāŻ¼├„┤_▓Īę“╝░╝ꎥ│╔åT’LļUĪŻ
▀zé„ĘĮ╩ĮĘų×ķ│Ż╚Š╔½¾w’@ąį▀zé„Īó│Ż╚Š╔½¾wļ[ąį▀zé„║═X▀Bµiļ[ąį▀zé„Ą╚ĪŻ│ŻęŖĄ─═╗ūā╗∙ę“░³└©PMP22Ż¼MPZŻ¼GJB1Ż¼MFN2ĪŻ│ŻęŖĄ─üåą═×ķCMT1Ż¼CMT2Ż¼CMTXĪŻCMT1×ķ│Ż╚Š╔½¾w’@ąį▀zé„Ą─├ō╦ĶŪ╩ąįCMTŻ¼ŲõųąCMT1A×ķūŅ│ŻęŖĄ─CMTüåą═Ż©š╝40Ī½50ŻźŻ®Ż¼Ųõ═╗ūā╗∙ę“×ķPMP22Ż╗CMT1Bš╝CMT1Ą─3Ī½5ŻźŻ¼Ųõ═╗ūā╗∙ę“×ķMPZĪŻX▀Bµiļ[ąį▀zé„Ą─CMTX1×ķĄ┌2│ŻęŖĄ─CMTüåą═Ż©š╝10ŻźŻ®Ż¼Ųõ═╗ūā╗∙ę“×ķGJB1ĪŻCMT2×ķ▌S╦„ąįCMTŻ¼Ųõųą│ŻęŖĄ─═╗ūā╗∙ę“░³└©MFN2Ż©š╝CMT2Ą─20ŻźŻ®ĪóMPZŻ©š╝CMT2Ą─5ŻźŻ®ĪóNEFL║═GDAP1Ą╚ĪŻCMT4×ķ│Ż╚Š╔½¾wļ[ąį▀zé„Ą─├ō╦ĶŪ╩ąįCMTŻ¼ŲõūŅ│ŻęŖĄ─═╗ūā╗∙ę“×ķGDAP1ĪŻPMP22ųžÅ═ĪóGJB1═╗ūāĪóPMP22╚▒╩¦ĪóMPZ═╗ūāĪóMFN2═╗ūā▀@5ĘNüåą═š╝╦∙ėąCMTĄ─92ŻźĪŻ
ŅA║¾Ż║ę“▓Ī│╠▀Mš╣ŠÅ┬²Ż¼ŅA║¾╔ą║├ĪŻ╝▓▓Ī║¾Ų┌┐╔─▄ć└ųžė░Ēæ╗ŅäėŻ¼Ą½║▄╔┘ī¦ų┬═Ļ╚½Üł╝▓Ż¼ę▓▓╗ė░Ēæš²│Żē█├³ĪŻ
ģó┐╝╬─½IŻ║
[1] Pareyson D, Marchesi C. Diagnosis, natural history, and management of Charcot-Marie-Tooth disease. Lancet Neurol, 2009,8:654-667.
[2] Rossor AM, Polke JM, Houlden H, Reilly MM. Clinical implications of genetic advances in Charcot-Marie-Tooth disease. Nat Rev Neurol, 2013,9:562-571.
[3] Saporta AS, Sottile SL, Miller LJ, et al. Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann Neurol, 2011,69:22-33.
░µÖÓ┬Ģ├„
ęį╔Žā╚╚▌üĒūį┴╝ßtģR-║▒ęŖ▓Īą┬▀Mš╣Ż¼╚ńėąĮ©ūh╗“ę╔å¢Ż¼ÜgėŁų┬ļŖ18017449015ĪŻ
 ķL░┤Č■ŠS┤a
ķL░┤Č■ŠS┤aĻPūóŠ½▓╩ā╚╚▌





