

一、基本信息
概述:先天性純紅細(xì)胞再生障礙性貧血是一種核糖體蛋白結(jié)構(gòu)基因突變導(dǎo)致核糖體生物合成異常的少見(jiàn)遺傳性疾病,又稱Diamond-Blackfan Anemia(DBA),是由紅細(xì)胞內(nèi)源性生成缺陷所致,呈常染色體顯性或隱性遺傳。絕大多數(shù)患兒起病發(fā)生于1歲以內(nèi),表現(xiàn)為大細(xì)胞性貧血、骨髓紅系細(xì)胞明顯減少、發(fā)育畸形和腫瘤易感性增高等。
病因:DBA于1936年由學(xué)者Josephs首次報(bào)道,1938年由學(xué)者Diamond和Blackfan報(bào)道描述,因而得名。DBA是一種核糖體合成障礙性疾病,是影響核糖體合成的基因突變所致。目前認(rèn)為核糖體功能缺陷引起選擇性紅系生成不良,表現(xiàn)為紅系定向祖細(xì)胞存在增殖、分化、凋亡及對(duì)細(xì)胞因子無(wú)反應(yīng)的內(nèi)在缺陷,因同時(shí)觀察到DBA患者有廣泛的軀體畸形,可以出現(xiàn)其他血細(xì)胞異常,發(fā)展為再生障礙性貧血,提示DBA也可累及除紅系造血外的多個(gè)系統(tǒng)。另外,核糖體的合成受損也影響腫瘤蛋白p53(tumor protein p53,TP53)腫瘤抑制通路的穩(wěn)定性和活性,這被認(rèn)為是疾病臨床表現(xiàn)(包括紅細(xì)胞生成受損、腫瘤易感)的原因,p53激活及下游事件導(dǎo)致細(xì)胞周期俘獲和細(xì)胞凋亡。通常表現(xiàn)為常染色體顯性遺傳,但是在一個(gè)家族中嚴(yán)重程度差異很大(外顯率降低)。
通過(guò)連鎖分析揭示DBA的遺傳基因位點(diǎn)已被陸續(xù)檢出。約25%的DBA患者是編碼核糖體蛋白19的基因(RPS19)發(fā)生突變。已有編碼核糖體大亞基基因(RPL35A、RPL5、RPL11)和核糖體小亞基基因(RPS24、RPS17、RPS7、RPS10、RPS26)致病性突變的描述,在50%以上的DBA患者中出現(xiàn)。其中RPL5 或 RPL11 突變患者更常出現(xiàn)軀體畸形。
DBA患兒EPO生成正常,尚無(wú)EPO抗體的描述。2012年研究發(fā)現(xiàn),DBA患者的造血轉(zhuǎn)錄因子GATA1也可出現(xiàn)剪接區(qū)突變,這種突變是除核糖體蛋白異常之外另一種可導(dǎo)致DBA的因素,可損害全長(zhǎng)蛋白(一種紅系分化所必需的轉(zhuǎn)錄因子)的生成,為X連鎖或常染色體隱性遺傳。其他的機(jī)制可能有原癌基因c-myc的激活。
流行病學(xué):由于DBA頗為少見(jiàn),其確切的發(fā)病率難以確定。歐洲回顧性研究表明,DBA在≤15歲兒童中年發(fā)病率約為1.5/100萬(wàn)~5.0/100萬(wàn)。此病發(fā)生于嬰幼兒,多數(shù)患兒出生后2周至2年發(fā)病,超過(guò)90%患兒在1歲內(nèi)確診,其中35%是在出生1個(gè)月內(nèi)診斷的。患兒多在就診時(shí)有明顯的貧血。本病男女患者之比約為1.1∶1,10%~25%有家族史。盡管大多數(shù)病例報(bào)道為高加索人,但是這種病例已在多種不同的人種中被發(fā)現(xiàn)。
二、疾病診斷
臨床表現(xiàn):
1.骨髓衰竭
35%患兒出生時(shí)即表現(xiàn)為貧血,常于生后2周至2年確診。貧血為大細(xì)胞或正細(xì)胞正色素性、網(wǎng)織紅細(xì)胞減少、白細(xì)胞正常或輕度降低、血小板正常或輕度增高、骨髓紅系增生低下而粒系和巨核細(xì)胞系增生活躍。
2.先天發(fā)育異常
30%~50%DBA患者可能存在先天發(fā)育異常,主要涉及頭部、上肢、心臟和泌尿生殖系統(tǒng)。如身材矮小,顱面部畸形包括顱面部過(guò)寬、寬平鼻梁、小頭、先天性白內(nèi)障、青光眼、斜視、硬腭高拱及唇腭裂甚至特納綜合征外貌,拇指畸形,先天性心血管發(fā)育異常,泌尿生殖器官畸形。存在身體的異常并不能預(yù)測(cè)患者血液系統(tǒng)疾病的嚴(yán)重程度。30%受累患者可出現(xiàn)生后的生長(zhǎng)遲緩,通常伴有其他先天畸形。然而,RPS19 基因連鎖突變則不會(huì)伴有生長(zhǎng)遲緩。
3.癌癥易感性增加
DBA患者腫瘤發(fā)生率約為4%,高于同年齡正常人群。且腫瘤發(fā)生年齡早,中位年齡為15歲。包括血液系統(tǒng)急性白血病、骨髓增生異常綜合征、淋巴瘤及實(shí)體瘤如骨肉瘤、胸腺癌、肝細(xì)胞癌、黑色素瘤、纖維組織細(xì)胞瘤、胃癌和腸癌等。患腫瘤后,較患同樣腫瘤的一般人群預(yù)后差,病死率高。
輔助檢查:
1.血常規(guī)及網(wǎng)織紅細(xì)胞計(jì)數(shù)檢測(cè)
對(duì)于疑似DBA者應(yīng)進(jìn)行,一般是大細(xì)胞性貧血,紅細(xì)胞平均容積(mean corpuscular volume,MCV)增大。通常在診斷時(shí)貧血很嚴(yán)重,2個(gè)月以下的嬰兒平均血紅蛋白水平65g/L,2個(gè)月以上的嬰兒為40g/L。然而,一些非典型的DBA患者僅有輕度貧血,或者僅有輕微的紅系異常。網(wǎng)織紅細(xì)胞計(jì)數(shù)明顯降低。通常患者的白細(xì)胞和血小板計(jì)數(shù)不受影響。
2.血紅蛋白電泳分析
因DBA患兒多表現(xiàn)為胎兒造血特征,6個(gè)月內(nèi)血紅蛋白F(hemoglobin F,HbF)百分比也比正常同齡兒升高,6個(gè)月后HbF仍持續(xù)升高,多保持在5%~10%,這些胎兒樣紅細(xì)胞特征表明應(yīng)激性紅細(xì)胞生成。
3.紅細(xì)胞中腺苷脫氨酶(erythrocyte adenosine deaminase,eADA)活性
患者通常存在嘌呤核苷代謝異常,可表現(xiàn)為47%~100%患者出現(xiàn)紅細(xì)胞腺苷脫氨酶的活性升高。
4.骨髓檢查
提示骨髓造血組織比例正常伴紅系前體細(xì)胞減少或缺失。90%以上患者的骨髓增生程度正常,僅早期紅系細(xì)胞明顯減少或缺如(多<5%),粒紅比例可達(dá)10:1,粒細(xì)胞系和巨核細(xì)胞系增生正常。
5.其他檢查
推薦進(jìn)行心臟超聲和腎臟影像檢查篩查器官發(fā)育異常可能。
診斷:
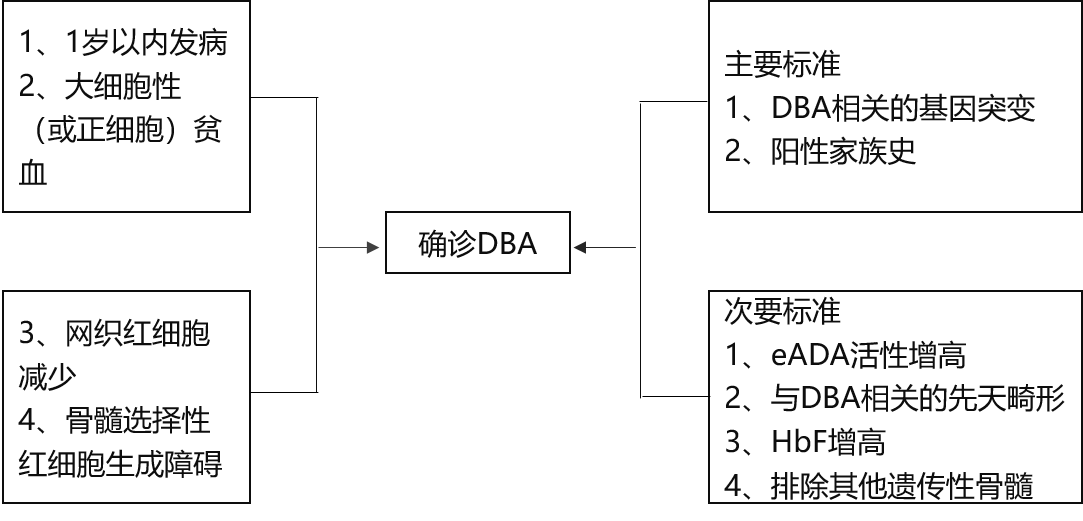
符合以下4條標(biāo)準(zhǔn)的患者可以診斷DBA:
1.發(fā)病年齡小于1歲。
2.大細(xì)胞性(或正細(xì)胞性)貧血,白細(xì)胞正常或稍降低、血小板正常或稍增高。
3.網(wǎng)織紅細(xì)胞明顯減少。
4.骨髓增生活躍伴紅系前體細(xì)胞明顯減少。
對(duì)于不滿足以上所有診斷標(biāo)準(zhǔn)的患者,可增加支持標(biāo)準(zhǔn),分為主要和次要發(fā)現(xiàn)以做出“擬診”。
1.主要支持標(biāo)準(zhǔn):包括存在與DBA相關(guān)的基因突變和陽(yáng)性家族史。
2.次要支持標(biāo)準(zhǔn):包括eADA活性增高、與DBA相關(guān)的先天畸形、HbF增高和排除其他遺傳性骨髓衰竭綜合征的證據(jù)。
在以下情況下可擬診DBA:
1.滿足3項(xiàng)診斷標(biāo)準(zhǔn)并有陽(yáng)性家族史。
2.滿足2項(xiàng)診斷標(biāo)準(zhǔn)和3項(xiàng)次要標(biāo)準(zhǔn)。
2.陽(yáng)性家族史和3項(xiàng)次要標(biāo)準(zhǔn)。
對(duì)于臨床高度懷疑DBA的患者,也建議進(jìn)行與DBA相關(guān)基因突變的基因篩查。最有效的方法是首先進(jìn)行RPS19 基因突變的序列分析(因?yàn)樵摶蛲蛔兪荄BA最常見(jiàn)的病因),然后通過(guò)分子學(xué)檢測(cè)方法檢測(cè)其他8個(gè)與DBA相關(guān)的基因。此外,如果患者具有DBA相關(guān)的基因突變,但不滿足DBA的診斷標(biāo)準(zhǔn),則可診斷為非典型DBA。
產(chǎn)前診斷:40%~50%的DBA患者為常染色體顯性遺傳,其余為散發(fā)的或者有不同遺傳特征的家族性。因?yàn)槭芟陆档耐怙@率及輕重兩種形式共存在同一家譜內(nèi)等因素的影響,DBA的遺傳規(guī)律很難預(yù)測(cè),即受多因素影響,表現(xiàn)型和基因型不一致。在DBA家族中應(yīng)進(jìn)行遺傳咨詢指導(dǎo)優(yōu)生優(yōu)育。
鑒別診斷:
1.兒童期暫時(shí)性幼紅細(xì)胞減少癥(transient erythroblastopenia of childhood,TEC)
TEC是兒童紅細(xì)胞生成減少最常見(jiàn)的病因,通常是由微小病毒B19感染引起的獲得性短暫的紅細(xì)胞生成不良。TEC表現(xiàn)為一過(guò)性的自身免疫介導(dǎo)的疾病,病程呈自限性,多發(fā)生于1歲以后,無(wú)陽(yáng)性家族史或先天畸形,于發(fā)病后1~2個(gè)月內(nèi)恢復(fù),預(yù)后良好。正細(xì)胞性貧血、血紅蛋白電泳進(jìn)行HbF含量和eADA正常、基因分析有助于鑒別TEC和DBA。
2.Aase綜合征
是以上肢畸形合并先天性紅系生成障礙為主要表現(xiàn)的綜合征。受累兒童也可能有心血管和顱面部(唇腭裂)畸形。此綜合征很可能是DBA的一種變異型而不是一種獨(dú)立的臨床疾病。與DBA一樣,一部分患者使用皮質(zhì)類固醇有效。
3.其他先天性骨髓衰竭性疾病
包括范科尼貧血、Shwachman-Diamond綜合征和先天性角化不良。此類疾病常伴有額外的血細(xì)胞減少,因表型復(fù)雜多變,基因輔助臨床診斷更為直接。
三、治療方式
治療:DBA的主要治療為皮質(zhì)類固醇和輸血。總體上,大約40%的DBA患者為類固醇依賴型,40%為輸血依賴型,20%到25歲時(shí)可獲得到緩解。10%~25%患者可自發(fā)緩解;約70%經(jīng)治療可達(dá)完全緩解,但仍有部分患者復(fù)發(fā);部分患者治療效果較差,主要靠輸血改善癥狀,故易引起血色病等。部分效果有限的治療選擇報(bào)道,如白介素-3、雄激素、甲氧氯普胺、免疫抑制劑如環(huán)孢素等。
1.糖皮質(zhì)激素
常用潑尼松2mg/(kg·d),以早晨單劑量一次或分兩次給藥。50%~70%患者初次治療時(shí)有效。越早治療,有效率越高,建議盡早接受治療。治療有效的患者通常在治療后1~2周即可出現(xiàn)網(wǎng)織紅細(xì)胞比例的升高。Hb上升至100g/L以上后可開(kāi)始減量。8~12周內(nèi)應(yīng)緩慢減少激素劑量直到達(dá)到最小有效劑量。這種最低有效劑量變化很大,并且許多對(duì)類固醇有反應(yīng)的患兒不能完全停藥,逐漸減量目標(biāo)為達(dá)到劑量≤0.5mg/(kg·d),目標(biāo)血紅蛋白為80~100g/L。患者仍在使用中等以上劑量激素治療時(shí),應(yīng)避免使用活病毒疫苗。治療應(yīng)嘗試進(jìn)行4周,如果無(wú)反應(yīng),則應(yīng)中止治療。類固醇無(wú)效者需警惕向骨髓衰竭發(fā)展,具有RPS26 基因突變的患者對(duì)類固醇治療的反應(yīng)率最低。
需要注意:在嬰兒中使用類固醇有嚴(yán)重的不良反應(yīng),尤其是早產(chǎn)兒中激素長(zhǎng)期應(yīng)用可能出現(xiàn)生長(zhǎng)延遲,學(xué)步時(shí)期易出現(xiàn)神經(jīng)肌肉發(fā)育不良及運(yùn)動(dòng)神經(jīng)延遲。所以多數(shù)專家推薦在6~12月齡之前應(yīng)避免使用類固醇治療,在此期間可以通過(guò)輸血治療進(jìn)行替代。對(duì)于長(zhǎng)期接受潑尼松治療的患者應(yīng)考慮開(kāi)始預(yù)防性使用磺胺甲惡唑-甲氧芐啶治療。
2.輸血治療
對(duì)于皮質(zhì)類固醇治療無(wú)效或存在類固醇使用禁忌的患者,輸血是主要治療手段。此類患者依賴長(zhǎng)期間斷性輸血,應(yīng)進(jìn)行完全紅細(xì)胞分型、去除白細(xì)胞處理,避免直系親屬供給。通常每4~6周輸血1次,血紅蛋白水平維持在80g/L以上,可保證生長(zhǎng)發(fā)育及日常活動(dòng)需要。
長(zhǎng)期輸血的患兒可引起鐵過(guò)載,應(yīng)監(jiān)測(cè)血清鐵蛋白,及時(shí)去鐵治療,建議去鐵治療可以在輸注懸浮紅細(xì)胞15次后或是患兒滿2歲后開(kāi)始。推薦地拉羅司20mg/(kg·d)起始,緩慢加量,最大劑量不超過(guò)40mg/(kg·d)。每3個(gè)月監(jiān)測(cè)血清鐵蛋白,目標(biāo)值為1000~1500μg/L。
3.基因治療
伴有RPS19 基因缺陷的DBA患者的基因治療正在研究中,體外實(shí)驗(yàn)顯示,在患者紅系祖細(xì)胞中增加RPS19 基因的表達(dá)可促進(jìn)紅細(xì)胞發(fā)育,用轉(zhuǎn)基因病毒載體來(lái)驗(yàn)證基因治療功效的動(dòng)物模型(去除RPS19 基因表達(dá))已經(jīng)被成功制成。
4.造血干細(xì)胞移植
類固醇無(wú)效、輸血依賴型DBA可以考慮造血干細(xì)胞移植。同胞供者需注意篩查排除供者攜帶致病基因突變。
診療流程圖:
先天性純紅細(xì)胞再生障礙性貧血診療流程
遺傳咨詢:40%~50%的DBA患者為常染色體顯性遺傳,其余為散發(fā)的或者有不同遺傳特征的家族性。因?yàn)槭芟陆档耐怙@率及輕重兩種形式共存在同一家譜內(nèi)等因素的影響,DBA的遺傳規(guī)律很難預(yù)測(cè),即受多因素影響,表現(xiàn)型和基因型不一致。在DBA家族中應(yīng)進(jìn)行遺傳咨詢指導(dǎo)優(yōu)生優(yōu)育。
參考文獻(xiàn):
[1] 張之南,郝玉書(shū),趙永強(qiáng),王建祥等.血液病學(xué).第2版.北京:人民衛(wèi)生出版社,2011:484-485.
[2] Willig TN, Gazda H, Sieff CA. Diamond-Blackfan anemia. Curr Opin Hematol,2000,7:85.
[3] Vlachos A, Ball S, Dahl N, et al. Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. Br J Haematol,2008,142:859.
[4] Vlachos A, Rosenberg PS, Atsidaftos E, et al. Incidence of neoplasia in Diamond Blackfan anemia: a report from the Diamond Blackfan Anemia Registry. Blood,2012,119:3815.
[5] Halperin DS, Freedman MH. Diamond-blackfan anemia: etiology, pathophysiology, and treatment. Am J Pediatr Hematol Oncol, 1989,11:380.
[6] Farrar JE, Nater M, Caywood E, et al. Abnormalities of the large ribosomal subunit protein, Rpl35a, in Diamond-Blackfan anemia. Blood, 2008,112:1582.
[7] Orfali KA, Ohene-Abuakwa Y, Ball SE. Diamond Blackfan anaemia in the UK: clinical and genetic heterogeneity. Br J Haematol, 2004, 125:243.
[8] Kuramitsu M, Sato-Otsubo A, Morio T, et al. Extensive gene deletions in Japanese patients with Diamond-Blackfan anemia. Blood, 2012, 119:2376.
[9] Chen S, Warszawski J, Bader-Meunier B, et al. Diamond-Blackfan anemia and growth status: the French registry. J Pediatr, 2005, 147:669.
[10] Abkowitz JL, Schaison G, Boulad F, et al.Response of Diamond-Blackfan anemia to metoclopramide: evidence for a role for prolactin in erythropoiesis. Blood, 2002,100(8):2687.
[11] Ball SE, Tchernia G, Wranne L, et al. Is there a role for interleukin-3 in Diamond-Blackfan anaemia? Results of a European multicentre study. Br J Haematol,1995,91(2):313.
[12] Leonard EM, Raefsky E, Griffith P, et al. Cyclosporine therapy of aplastic anaemia, congenital and acquired red cell aplasia. Br J Haematol,1989,72(2):278.
版權(quán)聲明
以上內(nèi)容來(lái)自良醫(yī)匯-罕見(jiàn)病新進(jìn)展,如有建議或疑問(wèn),歡迎致電18017449015。

 長(zhǎng)按二維碼
長(zhǎng)按二維碼關(guān)注精彩內(nèi)容





